INTRODUCCIÓN
Las anomalías oculares congénitas pueden presentarse aisladas y con frecuencia se asocian a otras alteraciones del desarrollo, que afectan a otros órganos o sistemas, como ocurre en los síndromes.(1,2)
El criptoftalmos u ojo oculto, también llamada ablefaria, es una anomalía muy poco frecuente, descrita clínicamente por Zehender en 1872, y desde el punto histopatológico por Manz en el mismo año. Se estima en, aproximadamente, 20 por cada 100 000 nacidos vivos y se presenta como la ausencia congénita de los párpados, los ojos están cubiertos por piel que va desde la frente hasta las mejillas y ocasiona una pérdida de la arquitectura total o parcial de las estructuras del globo ocular. Básicamente el defecto radica en la ausencia de fisura palpebral. Hay ausencia en grado variable de pestañas y cejas. Con frecuencia es bilateral y simétrica,(1,2,3,4,5) aunque, hay casos unilaterales y asimétricos.(6)
El trabajo realizado pretendió describir la etiología, incidencia y características, debido a lo poco frecuente de la aparición de esta entidad, y para una tener una mejor comprensión y elevar la calidad de la atención de estos pacientes y las gestantes.
PRESENTACIÓN DEL CASO
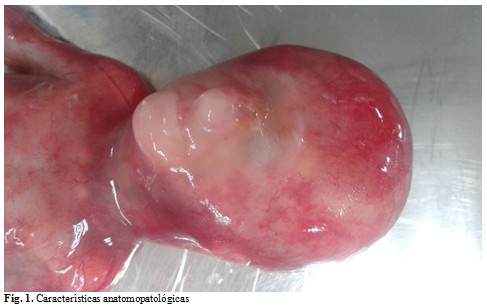
Se presenta el caso de una gestante de 30 años de edad, sin antecedentes patológicos personales. En la ecografía de las 17 semanas el equipo multidisciplinario de genética médica provincial observó disgenesia del segmento anterior y se le brindó asesoramiento genético. A la edad gestacional de 14 semanas la pareja decidió interrumpir el embarazo y se le realizó necropsia clínica al cuerpo fetal en el Hospital General Universitario Dr. Gustavo Aldereguía Lima, de Cienfuegos. En el habitus exterior se estudió un feto masculino con ausencia total de ambos párpados, donde la piel recubría toda la superficie hasta las mejillas. Ambos globos oculares presentaban pérdida total de su arquitectura, además, la ausencia de orificios nasales, auditivos y escaso desarrollo del cartílago auricular de ambas orejas con implantación baja. En el examen interno no se encontraron alteraciones. Las conclusiones anatomopatológicas en este caso fueron: criptoftalmo bilateral completo, atresia de orificios nasales y auditivos, hipoplasia de ambos cartílagos auriculares e implantación baja de las orejas. (Fig. 1)
DISCUSIÓN
Se plantea la herencia autosómico recesiva aunque existen reportes de casos con presentación autosómica dominante sin predilección por sexo o raza.(1,2,5)
La etiología del criptoftalmos se desconoce, solo se ha sugerido la deficiencia de vitamina A, la presencia de bandas amnióticas,(1,2,5) pero en este caso que se presenta, no se recogieron estos datos.
Se origina por un defecto congénito en la migración de la cresta neural que da lugar a un desarrollo anormal de los párpados y de las estructuras oculares anteriores; incluye la ausencia parcial o total de las cejas, hendidura palpebral, pestañas y conjuntiva. Pueden existir los ojos ocultos debajo de la piel y por lo general son disfuncionales. En muchas ocasiones aparece un esbozo de las cejas y de ahí la piel continúa de la ceja a la mejilla; el resto de la órbita y de la cara suelen ser normales.(1) El caso presentó ausencia total de hendidura palpebral, conjuntivas y distorsión total de la arquitectura de ambos globos oculares. Todo esto acompañado de atresia de orificios nasales y auditivos, hipoplasia de ambos cartílagos auriculares e implantación baja de ambas orejas. No fue posible evaluar la formación de cejas y pestañas por la edad gestacional fetal en el momento de la autopsia.
Esta anomalía puede ubicarse en el segundo mes de vida intrauterina. Alrededor de la séptima semana aparecen unas invaginaciones laterales y ocurre a nivel de la copa óptica un levantamiento ectodérmico por debajo del mesodermo que la rodea que forma un par de pliegues (párpados) que tienden a aproximarse hasta quedar fusionados alrededor de la novena semana, pero a partir del quinto mes del desarrollo esta unión comienza a desintegrarse, aunque no se reabre totalmente hasta el séptimo u octavo mes del desarrollo; si no ocurre esta cascada de eventos embriológicos de forma ordenada, se presenta el criptoftalmos o bien alteraciones a nivel palpebral, según sea la intensidad de la noxa.(1,5) En este caso, a pesar que la interrupción del embarazo se realizó a las 17 semanas, no se identificaron ningunas de las estructuras.
Zehender y Manz clasificaron la criptoftalmia en tres subgrupos:
- Típico: el más frecuente. La piel reemplaza a los párpados. No existen cejas ni pestañas. Se corresponde con el caso presentado.
- Parcial: la piel se fusiona en la zona medial del globo. El tercio externo del párpado es normal.
- Congénito: simbléfaron del párpado superior con el globo. La córnea aparece queratinizada, el punto lagrimal está ausente y puede aparecer xerosis ocular.(6)
Otros rasgos oftalmológicos asociados pueden ser: microftalmos, atalamia, ausencia de malla trabecular y canal de schlem, subluxación del cristalino, aniridia, sinequías de iris y cristalino al endotelio corneal, atrofia del cuerpo ciliar, colobomas coroideos, cejas supernumerarias, ausencia de folículos pilosos y glándulas lagrimales.(6) Ninguno de estos rasgos se corresponde con los del caso presentado.
Hallazgos no oftalmológicos descritos: disencefalia, mielomeningocele, retraso mental, sindactilia, hernia umbilical, atresia anal, aplastamiento frontotemporal, distribución capilar anormal, malformaciones otorrinolaringológicas, genitourinarias o cardíacas.(6,7) De este grupo de rasgos, el caso presentado mostró atresia de orificios nasales y auditivos, hipoplasia de ambos cartílagos auriculares e implantación baja de ambas orejas.
Pueden existir cambios metaplásicos desde el epitelio corneal hasta la piel. Los músculos orbicular y elevador del párpado superior se encuentran bien representados, mientras que el tarso y la conjuntiva son rudimentarios o no se encuentran.(6)
Thomas y cols. en su artículo: Isolated and syndromic cryptophthalmos, publicado en la revista American Journal of Medical Genetics, número 25 de 1986, propusieron una serie de criterios mayores y menores, lo cual permitió distinguir entre la criptoftalmia aislada de la sindrómica no necesariamente incluida en el síndrome de Fraser (SF); según refieren Caoa y cols. en su artículo acerca del tema, sin embargo, son escasas las descripciones histopatológicas en el criptoftalmos y se realizaron en el contexto del SF o en sus modelos animales, que sugirieron alteraciones de la matriz extracelular.(4)
Para el manejo de las malformaciones congénitas oculares se requiere asesoramiento genético con el objetivo de brindar información útil a los padres y orientarlos sobre el riesgo de recurrencia en embarazos posteriores,(1) también podrá plantearse la posibilidad de la cirugía reconstructiva en casos complejos y solo si hay resultados de potenciales evocados visuales confirmados por pruebas electrofisiológicas.(1,8)
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Contribución de los autores:
1. Conceptualización: Lilian Rachel Vila Ferrán.
2. Curación de datos: Lilian Rachel Vila Ferrán, Katia Rodríguez Palacios, Leydi María Sosa Águila.
3. Análisis formal: Lilian Rachel Vila Ferrán, Katia Rodríguez Palacios, Leydi María Sosa Águila.
4. Adquisición de fondos: Esta investigación no contó con la adquisición de fondos.
5. Investigación: Lilian Rachel Vila Ferrán, Katia Rodríguez Palacios, Leydi María Sosa Águila.
6. Metodología: Leydi María Sosa Águila.
7. Administración del proyecto: Lilian Rachel Vila Ferrán.
8. Recursos: Lilian Rachel Vila Ferrán, Katia Rodríguez Palacios, Leydi María Sosa Águila.
9. Software: Katia Rodríguez Palacios.
10. Supervisión: Katia Rodríguez Palacios.
11. Validación: Leydi María Sosa Águila.
12. Visualización: Lilian Rachel Vila Ferrán.
13. Redacción del borrador original: Lilian Rachel Vila Ferrán, Katia Rodríguez Palacios, Leydi María Sosa Águila.
14. Redacción, revisión y edición: Lilian Rachel Vila Ferrán, Katia Rodríguez Palacios, Leydi María Sosa Águila.
REFERENCIAS BIBLIOGRÁFICAS
1- Bauza FY, Góngora TJ, Veitía RZ, Rojas RI. Criptoftalmos bilateral. Rev Cubana Oftalmol[Internet]. 2016[citado 24/3/25];29(2):[aprox. 10p.]. Disponible en: https://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-21762016000200014.
2- Moore KL, Persaud TV, Torchia MG. Desarrollo de los ojos y los oídos. En: Moore KL, Persaud TV, Torchia MG. Embriología clínica[Internet]. 9na ed. Madrid:Elsevier;2013[citado 15/9/2024]. Disponible en: https://www.academia.edu/37872323/Embriologia_Clinica_9a_Edicion_Moore.
3- American Academy Ophthalmology. Fundamentos y Principios de Oftalmología. Barcelona:Elsevier;2008.
4- Cao G, Navacchia D, Zárate JO. Criptoftalmos unilateral no sindrómico. An Pediat[Internet]. 2008[citado 24/3/25];68(2):[aprox. 2p.]. Disponible en: https://www.analesdepediatria.org/es-criptoftalmos-unilateral-no-sindromico-articulo-S1695403308749166.
5- González TJ, Salcedo CG, Villanueva MC, García GJ. Criptoftalmos y ablefarón. Presentación de un caso. Rev Mex Oftalmol[Internet]. 2008[citado 24/3/25];82(3):[aprox. 3p.]. Disponible en: https://www.medigraphic.com/pdfs/revmexoft/rmo-2008/rmo083h.pdf.
6- Bauza Y, Góngora JC, Veitía ZA, Rojas I. Rev Cubana Oftalmol[Internet]. 2016[citado 24/3/25];29(2):[aprox. 4p.]. Disponible en: https://revoftalmologia.sld.cu/index.php/oftalmologia/article/view/392/html_218.
7- Flores G, Pérez T, Pérez M. Malformaciones congénitas diagnosticadas en un hospital general. Revisión de cuatro años. Act Pediatr Mex[Internet]. 2011[citado 24/3/25];32(2):[aprox. 6p.]. Disponible en: https://www.medigraphic.com/pdfs/actpedmex/apm-2011/apm112d.pdf.
8- Harraquian JV, Brandão FM, Dos Santos GK, Gonçalves RM. Criptoftalmia unilateral: relato de caso[Internet]. Florianápolis:3er Congreso Internacional y Oculoplastia;2024[citado 24/3/25]. Disponible en: https://atenaeditora.com.br/catalogo/dowload-post/92800.
