INTRODUCCIÓN
La enfermedad de Wilson (EW) constituye un problema de salud mundial. Es un trastorno hereditario descrito por primera vez por Samuel Alexander Kinnear Wilson en 1912. Si no se atiende de forma adecuada, provoca lesiones irreversibles en el hígado y en el cerebro, que pueden conducir a la muerte en edad pediátrica. Se caracteriza por la acumulación de cobre en hígado, cerebro, córnea y otros tejidos.(1) Las manifestaciones clínicas presentes en los pacientes con esta enfermedad son muy variables, lo cual contribuye a las dificultades actuales1en el diagnóstico clínico.(1,2,3,4,5)
Se informan pocos casos con presencia de trastornos en la coagulación, sin embargo, su estudio es de interés, debido a las consecuencias que acarrea.(6,7,8) Para el estudio de quienes padecen de EW es necesario un equipo multidisciplinario constituido por gastroenterólogos, hepatólogos, genetistas, bioquímicos, neurólogos, psiquiatras, nefrólogos, oftalmólogos, en dependencia de la sintomatología, y si se trata de alteraciones hematológicas se incluye al hematólogo.
La causa molecular que provoca la EW son las mutaciones en el gen atp7b, de las cuales en la actualidad se han informado más de 700.(9,10,11,12,13)
Este trabajo tiene como objetivo identificar los trastornos de la coagulación y su causa molecular en pacientes cubanos con diagnóstico clínico de enfermedad de Wilson.
MÉTODOS
Se llevó a cabo un estudio descriptivo transversal, con 50 pacientes cubanos con enfermedad de Wilson, cuyo diagnóstico clínico fue realizado en el Instituto de Gastroenterología; y el diagnóstico molecular en el Centro Nacional de Genética Médica, durante el período 2011-2013. Los pacientes dieron su consentimiento por escrito para participar en la investigación, mediante un documento elaborado para tal fin.
Se extrajo el ADN de la sangre de los pacientes por el método de precipitación salina. La amplificación de los fragmentos de interés se hizo mediante la técnica PCR (Reacción en Cadena de la Polimerasa); y la búsqueda de mutaciones en los exones 2, 3, 6, 8, 10 y 14 del gen atp7b, mediante las técnicas SSCP (Polimorfismo Conformacional de Simple Cadena), digestión enzimática y secuenciación. Esta investigación fue aprobada por el Comité de Ética y Consejo Científico del Centro Nacional de Genética Médica.
Las variables analizadas fueron las que caracterizaron la muestra en general, como sexo, edad, edad al inicio de la enfermedad, edad al momento del diagnóstico; y otras como: presencia de trastornos de coagulación, presencia de cambios conformacionales y presencia de la mutación p.L708P en el exón 8 del gen atp7b.
Detección de los cambios conformacionales mediante la técnica de SSCP
Se mezcló 3,5μl con una solución de parada de bromofenol azul (0,05 % BFA, 10mM NaOH, 95 % formamida, 20mM EDTA) y 1μL del producto amplificado, en un volumen final de 7μL. Se aplicó en un gel de acrilamida comercial (GeneGel Excel 12,5/24 Kit). La visualización del ADN se realizó por el método de tinción con plata, siguiendo las instrucciones del juego comercial kit PlusOne DNA Silver Staining (Amersham Biosciences, 2007).
Visualización del ADN con el método de tinción con plata
Después de las corridas electroforéticas de las muestras con el uso de la técnica de SSCP, la visualización del ADN se realizó por el método de tinción con plata. Se siguieron las instrucciones del juego comercial PlusOne DNA Silver Staining (Amersham Biosciences, EUA).
Detección de la mutación p.L708P en el exón 8 del gen atp7b
Se amplificó el exón 8 del gen atp7b y se procedió a la búsqueda de la mutación p.L708P. Se digirió el producto de amplificación con la enzima de restricción Alu I, a 37 °C, durante tres horas. Se realizó la digestión en un volumen final de 30 μL con 15 μL del producto que se amplificó, y 20 U de la enzima Alu I.
Electroforesis y visualización de los fragmentos digeridos
Para la separación de los fragmentos que se digirieron se realizó la electroforesis en el gel de agarosa al 2 % para la mutación p.L708P. El voltaje que se usó fue de 250 V y el gel fue teñido con bromuro de etidio al 5X. Se utilizó el tampón TBE 10X (0,5 % Tris, 10 mM EDTA, 0,5 % ácido bórico) para la corrida electroforética. El ADN se visualizó por exposición a la luz ultravioleta en el transluminador 4000 (Stratagene, EUA).
Análisis del impacto funcional de la mutación p.L708P en la proteína ATP7B
Se utilizó el programa Polyphen-2 para analizar el efecto de la mutación p.L708P en la proteína ATP7B.(14)
RESULTADOS
Del total de pacientes analizados, 18 fueron del de sexo femenino (36 %), y 32 de sexo masculino (64 %). La edad media de inicio de la EW en los pacientes analizados (29,8 años ±7,8). La edad de diagnóstico fue de 31,1 años ±3,5).
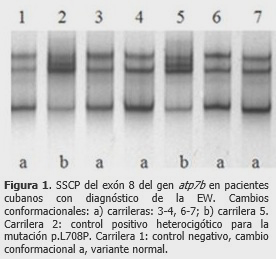
Se detectó una paciente de 14 años con trastornos de los factores de la coagulación, encefalopatía hepática, irregularidad menstrual, problemas del crecimiento y depresión; manifestaciones clínicas que tuvieron una rápida aparición. Al realizar el análisis del exón 8, se visualizaron dos cambios conformacionales diferentes a la variante normal, denominados a y b. (Figura 1).
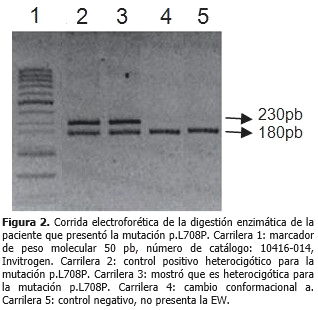
La corrida electroforética de esta paciente que presentó los cambios conformacionales b, fue similar al patrón electroforético del control positivo heterocigótico para la mutación p.L708P. Se mostró una paciente con cambio conformacional b (2 %). (Figura 2).
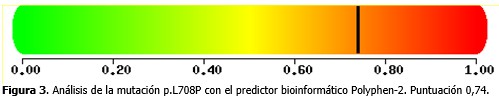
El análisis de la mutación p.L708P mediante el programa Polyphen-2 mostró una puntuación de 0,74, lo que significó que puede provocar daño a la proteína ATP7B. (Figura 3).
DISCUSIÓN
Un paso previo a la búsqueda de las mutaciones en el gen atp7b, es la detección de cambios conformacionales por la técnica de SSCP, y en esta investigación el cambio conformacional b correspondió a la mutación p.L708P.
La técnica de la digestión enzimática permitió confirmar la presencia de la mutación p.L708P. En la electroforesis se mostró el control heterocigótico positivo para esta, el cual presentó el sitio de restricción para la enzima AluI en uno de sus alelos, fragmento 230pb. El paciente que tuvo el cambio conformacional a, presentó las bandas, 180pb y 50pb, el último fragmento no se encontró en la electroforesis, pues migró con el frente de corrida. La única paciente con trastornos de los factores de la coagulación exhibió los fragmentos 230pb, 180pb y 50pb, por lo que resultó heterocigótica para la variante p.L708P.
La mutación p.L708P es la variante más frecuente en Islas Canarias(15) y se ha documentado una frecuencia de 2 % en pacientes cubanos.(16) La paciente que resultó heterocigótica para esta mutación, refirió que su bisabuela materna procedía de las Islas Canarias; ello evidencia la importancia de considerar datos como este de la ascendencia de los pacientes, en los estudios genéticos.
La mutación p.L708P provoca la sustitución de timina por citosina en la posición 2123 del gen atp7b. Se localiza en el segundo segmento de transmembrana de la proteína ATP7B, la cual tiene la función de transportar el cobre en el hepatocito.
La frecuencia de la presencia de trastornos de los factores de la coagulación en esta serie de 50 pacientes con diagnóstico presuntivo de EW fue de 2 %, muy baja si se compara con lo que se informa en la literatura.(6,17,18) Es válido destacar que en la paciente con dichos trastornos, se identificó la causa molecular de la enfermedad, la mutación p.L708P. Esto es fundamental para la identificación de portadores y pacientes asintomáticos en la familia, y realizar acciones de prevención. En su tratamiento participaron un conjunto de especialistas, tales como, gastroenterólogos, genetistas, neurólogos, hepatólogos, bioquímicos, hematólogos, entre otros. La EW, aunque no es una enfermedad hematológica, puede asociarse a trastornos de los factores de la coagulación, lo que en algunos casos requiere de reorientar el tratamiento en beneficio del paciente.
La otra mutación causante de la enfermedad no fue identificada en los exones analizados, (2, 3, 6, 8, 10 y 14). Por tal motivo, debe ampliarse el estudio molecular en la paciente, para examinar los restantes exones del gen atp7b. Sin embargo, la identificación de la mutación p.L708P fue crucial para el tratamiento y seguimiento.
Se concluye que la frecuencia de aparición de los trastornos de los factores de la coagulación en pacientes cubanos con diagnóstico clínico de EW, es baja respecto a la informada por otros estudios, realizados en otras poblaciones. Sin embargo, hay que considerarla como una probabilidad real, y realizar las pruebas necesarias para su confirmación, dadas las implicaciones que tiene para la salud de estos pacientes. La identificación de la causa molecular en la paciente permitió la confirmación de la EW.
Los autores agradecen a la técnica Lídice Reyes y a la DrC. Teresa Collazo, por su contribución a la investigación.
Conflicto de interés: No existe conflicto de interés.
Contribución de los autores: Revisión de la literatura: Liudmila Feoktistova Victorova, Caridad Ruenes Domech, Elsa F García Bacallao, Hilda Roblejo Balbuena, Estela Morales Peralta, Yulia Clark Feoktistova; diagnóstico clínico: Caridad Ruenes Domech, Elsa F García Bacallao, Hilda Roblejo Balbuena, Estela Morales Peralta; diagnóstico molecular: Yulia Clark Feoktistova; escritura del artículo: Liudmila Feoktistova Victorova, Yulia Clark Feoktistova.
Financiación: Centros implicados en la investigación: Centro Nacional de Genética Médica y Instituto Gastroenterología.
