INTRODUCCIÓN
El síndrome de Marfan (SM) es una enfermedad genética autosómica dominante del tejido conectivo, caracterizada por una combinación variable de manifestaciones cardiovasculares, músculo-esqueléticas y oftalmológicas.(1,2)
En la inmensa mayoría de los casos se debe a mutaciones en el gen FBN1, ubicado en el cromosoma 15q21.1; este codifica para la fibrilina-1,(2,3) proteína esencial del tejido conectivo, dando origen a un defecto hereditario que ocasiona una formación de fibras elásticas anormales, con la consiguiente disfunción de los tejidos que la poseen.(4) Por otro lado, se ha reportado que puede ser causada por mutaciones en el gen TGF- BR2, el cual se localiza en 3p 24.2-25.(5,6)
Esta identidad fue descrita por primera vez en el año 1896 por el médico francés Antoine Bernard-Jean Marfan;(6,7) y ha recibido desde entonces varios nombres: aracnodactilia (Achard, 1902); hipercondroplasia (Méry y Babonneix, 1902); distrofia mesodérmica congénita (Weber); o dolicostenomelia.(8) Fue incluida en 1955 en una clasificación de enfermedades del tejido conectivo. En 1986 un panel internacional de expertos definió un conjunto de criterios clínicos (nosología de Berlín) para su diagnóstico, con una modificación posterior en 1996, referida desde entonces como nosología de Gante. Esta última, que incluyó la presencia de mutaciones en el gen FBN1, y unos criterios más restrictivos que los de la nosología de Berlín, tuvo como objetivos disminuir el sobre diagnóstico del síndrome y facilitar mejores guías para diferenciarlo de otras entidades que se superponían.(7) Actualmente el diagnóstico se basa en los Criterios Gante revisados para diagnóstico de síndrome de Marfan enunciados en 2009.(2)
Los síntomas pueden aparecer a cualquier edad y son muy variables, incluso dentro de una misma familia.(5) Entre las manifestaciones cardiovasculares destaca la afectación mitral y de los grandes vasos; la afección aórtica es la que presenta mayor impacto pronóstico.(9) Habitualmente, la afectación esquelética es el primer signo de la enfermedad.(10) La afectación oftalmológica más frecuente es la subluxación del cristalino, sin embargo se presentan otras como córnea plana, pupila con pobre respuesta a la dilatación farmacológica, aumento del eje anteroposterior y miopía moderada.(5)
La incidencia es de uno por cada 3 000 a 5 000 nacimientos; es de las más altas entre los trastornos hereditarios.(3) En la mayor parte de los grupos raciales y étnicos, sin diferenciación de sexo, cerca del 90 % de las mutaciones documentadas son de tipo único y afectan a un individuo o familia; el 20 % de los pacientes no heredan las mutaciones, por lo que estas se interpretan como nuevas.(2)
Según los Criterios Gante revisados se otorga mayor peso a tres componentes: 1) hallazgos clínicos de aneurisma/disección aórtica y ectopia del lente ocular; 2) pruebas genéticas del FBN1 y otros genes como TGFBR1 y 2; y 3) un énfasis diagnóstico del SM hacia una diferenciación de otras entidades.(2)
El diagnóstico de SM puede ser relativamente fácil si se tienen suficientes datos oculares, esqueléticos y/o cardiovasculares. Sin embargo, cabe mencionar que se observa expresividad variable tanto dentro de una familia como entre diferentes familias; además, existen otras entidades genéticamente determinadas con un hábito semejante, y en la población general se pueden encontrar con cierta frecuencia individuos altos y con miopía, datos aislados con los que no se puede establecer un diagnóstico.(5,10) El presente informa tiene como objetivo exponer el caso clínico de una mujer adulta con SM.
PRESENTACIÓN DEL CASO
Se presenta el caso de una paciente del sexo femenino, piel blanca, de 44 años de edad y de procedencia urbana (Cumanayagua); debido a antecedentes patológicos personales de luxación bilateral del cristalino, había sido operada del ojo derecho; también refirió desprendimiento de retina del ojo izquierdo hacía cuatro años aproximadamente, miopía y escoliosis. Sin antecedentes patológicos familiares, ni hábitos tóxicos.
Acudió al Cuerpo de Guardia del Hospital Provincial Gustavo Aldereguía Lima, de Cienfuegos, refiriendo cansancio a pequeños esfuerzos, acompañado de dificultad al respirar y falta de aire al caminar, además de sensación de opresión torácica. En el examen cardiovascular presentó cifras elevadas de tensión arterial (170/100mmHg). Ante estos elementos se decidió su ingreso bajo la impresión diagnóstica de hipertensión arterial descompensada.
En interrogatorio más detallado en la sala, se conoció el antecedente de dolor dorsolumbar, de moderada intensidad, continuo, de aproximadamente una semana de evolución.
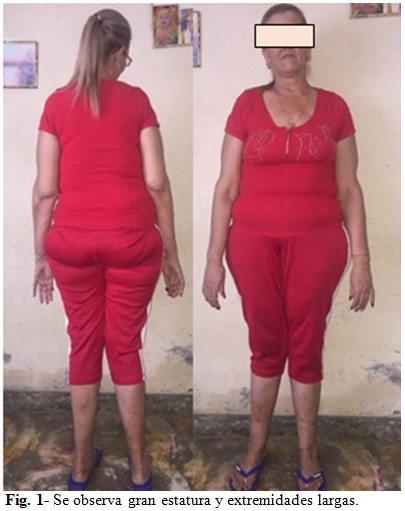
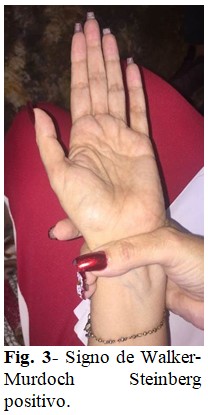
A simple vista se apreció gran estatura, extremidades largas (Fig. 1), presencia de aranodactilia, con signos de Steinberg (Fig. 2) y Walker-Murdoch (Fig. 3) positivos; así como dedos largos en miembros inferiores. (Fig. 4). Además, se encontró estrabismo divergente, sumándosele también una cifoescoliosis manifiesta.


Se le realizaron exámenes complementarios; los de laboratorio resultaron entre los límites de la normalidad; el electrocardiograma aportó signos de sobrecarga ventricular izquierda y arritmia extrasistólica; la radiografía posteroanterior de tórax informó mediastino ensanchado de aspecto vascular. Por su parte, el ecocardiograma informó los siguientes resultados:
- Septum interventricular: 14 mm.
- Diámetro del ventrículo izquierdo: 67 mm.
- Pared posterior: 12 mm.
- Fracción de eyección del ventrículo izquierdo: 45 %.
- Aurícula izquierda: 25 mm.
- Anillo aórtico: 24 mm.
- Raíz aórtica: 32 mm.
- Aorta ascendente: 36 mm.
- No se precisó colgajo intimal.
- Patrón de relajación diastólica prolongado y fracción segmentaria global ligeramente deprimida.
- Insuficiencia aórtica de moderada a severa con área de 8 cm.
- Insuficiencia mitral ligera moderada con área de 4,13 cm.
- A nivel de arco aórtico se observó imagen de flap íntima y doble luz; además de aorta descendente dilatada con doble luz.
La suma de los elementos clínicos y ecocardiográficos orientó hacia el diagnóstico de síndrome de Marfan, por lo que fue evaluado el caso de conjunto con el servicio de Cardiología y Angiología. Se decidió su traslado al Cardiocentro de Villa Clara, donde se le realizó angioTAC y se observó disección aórtica posterior a la emergencia de la arteria subclavia izquierda que recanalizaba antes del tronco celiaco, a partir de la cara dorsal por delante, y a la izquierda de la mesentérica superior volvía a disecarse respetando ambas arterias renales, extendiéndose hasta ambas iliacas externas.
El caso fue nuevamente discutido en colectivo con cirujanos vasculares, y se decide diferir manejo quirúrgico por la extensión de la lesión. Se enfocó el tratamiento hacia el control farmacológico.
La paciente actualmente tiene estabilidad clínica. Recibe tratamiento farmacológico, incluyendo betabloqueadores. Se mantiene en seguimiento por consulta externa y se realiza vigilancia de las complicaciones.
DISCUSIÓN
El SM es una enfermedad hereditaria que generalmente no se manifiesta en edades tempranas; ya en la adolescencia y después de los 20 años se empiezan a observar las alteraciones esqueléticas, algunas oculares, como la disminución temprana de la agudeza visual y los síntomas cardiovasculares.(3)
A pesar de tratarse de una enfermedad autosómica dominante, existe un porcentaje (5-35 %) de los casos en los cuales aparece de forma esporádica y en los cuales se pueden presentar nuevas mutaciones cromosómicas que generen una expresividad diferente de la enfermedad.(3) En ese grupo de pacientes se encuentra el caso presentado, pues no se recogen antecedentes familiares, o al menos los familiares no presentan rasgos fenotípicos distintivos de la enfermedad.
Este síndrome se diagnostica fundamentalmente a partir de los criterios de Gante revisados,(11) en los cuales se da más peso a las alteraciones de la raíz aórtica y ectopia lentis.
En ausencia de antecedentes familiares de la enfermedad, se puede establecer el diagnóstico de la enfermedad de Marfan cumpliendo uno de los siguientes criterios:
- Criterio aórtico (diámetro de la línea Z mayor que 2 o igual, o disección aórtica) y ectopia lentis.
- Criterio aórtico y una mutación del FBN 1.
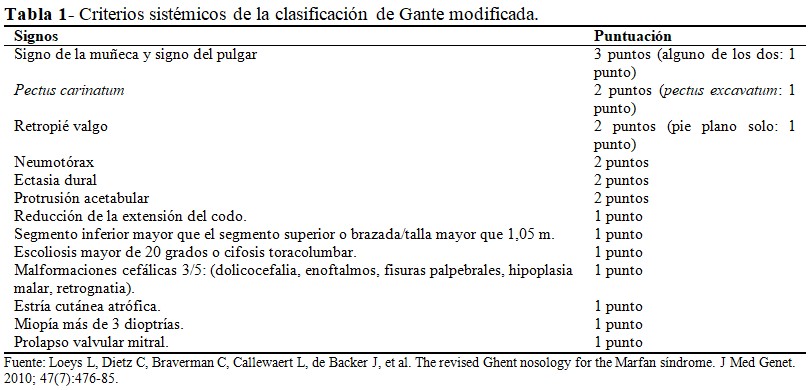
- Criterio aórtico y 7 puntos o más en la puntuación de manifestaciones sistémicas de la escala de Gante revisada (Tabla 1).
- Presencia de ectopia lentis y mutación del FBN 1 que se ha identificado con la lesión aórtica aneurismática.
Si existen antecedentes de la enfermedad, mediante la presencia de alguno de estos criterios se establece el diagnóstico:
- Ectopia lentis.
- Siete puntos o más de manifestaciones sistémicas en la escala de Gante revisada (Tabla 1).(11)
- Criterio aórtico.
Los criterios Gante han sido mundialmente utilizados en el diagnóstico del SM, con una altísima especificidad, al haberse detectado mutaciones en FBN1 hasta en el 97 % de los pacientes que reúnen estos criterios.(12) Sin embargo, Cabrera y colaboradores(7) señalan sus limitaciones, tales como no considerar la dependencia de la edad en algunas manifestaciones clínicas (lo que dificulta el diagnóstico en niños), o incluir algunas manifestaciones físicas no específicas o con valor diagnóstico escasamente validado. Esto puede dar lugar a diagnosticar erróneamente SM a pacientes con síndrome de ectopia lentis, síndrome de prolapso de válvula mitral, o por el contrario, no hacerlo en pacientes con ectopia lentis y dilatación aórtica sin suficientes manifestaciones esqueléticas.(7)
Luego de la revisión de los criterios establecidos y teniendo en cuenta la ausencia de antecedentes familiares de la enfermedad en el caso que se presenta, se puede establecer el diagnóstico de la enfermedad de Marfan cumpliendo con el criterio aórtico y ectopia lentis; pues la paciente presenta descripción ecocardiográfica (aorta descendente dilatada con doble luz) y tomográficas compatibles con disección aórtica: disección aórtica posterior a la emergencia de la arteria subclavia izquierda que recanaliza antes del tronco celiaco, a partir de la cara dorsal por delante, y a la izquierda de la mesentérica superior vuelve a disecarse respetando ambas arterias renales, extendiéndose hasta ambas iliacas externas; asimismo, se recoge antecedente de luxación bilateral del cristalino. Por otra parte, suma 9 puntos de acuerdo a la escala de afectación sistémica: signo de la muñeca y signo del pulgar (3 puntos), pie plano (1 punto), reducción de la extensión del codo (1 punto), malformaciones cefálicas (1 punto), segmento inferior mayor que el segmento superior o brazada/talla mayor que 1,05 m (1 punto), escoliosis o cifosis toracolumbar (1 punto), y miopía más de 3 dioptrías (1 punto).
Se describe como criterio imprescindible la presencia de los signos de Steinberg y Walker-Murdoch; el primero consiste en la protrusión del pulgar en oposición forzada más allá del borde cubital de la mano, y el segundo es evidente al superponerse los dedos pulgar y meñique en más de 1-2 cm al hacer prensión de la muñeca, proximalmente a la apófisis estiloides del radio con la otra mano,(11,12) signos que fueron encontrados en la paciente.
La totalidad de las fuentes revisadas señalan que la complicación de mayor severidad e índice de mortalidad es precisamente la disección aórtica, resultado de un diagnóstico tardío de la enfermedad, afección que en esta paciente logra extenderse hasta las porciones iliacas de la aorta, siendo riesgosa la corrección quirúrgica, lo que traduce un estado avanzado de dicha complicación. La ecocardiografía transtorácica o transesofágica permite realizar el diagnóstico y evaluar la progresión de las lesiones cardiovasculares, a la vez que determina el momento oportuno para una opción quirúrgica,(13) sin embargo, como se evidenció, se requiere la realización de otros estudios imagenológicos para determinar la extensión de la afectación aórtica.
La dilatación aórtica puede conducir a una insuficiencia de la válvula aórtica e insuficiencia de la mitral; el daño valvular más frecuente en estos pacientes es el prolapso de la válvula mitral,(12) sin embargo este caso presentaba insuficiencia mitral de ligera a moderada. En este sentido, en estudio realizado por Soto y colaboradores(14) se evidenció prolapso valvular mitral en 67 % de los pacientes, e insuficiencia mitral en 45 %, por lo que la frecuencia de la insuficiencia mitral no resulta despreciable.
En estudio realizado por Borulu y Erkut(15) en pacientes operados de aneurisma de aorta ascendente, la causa más frecuente fue la ectasia anuloaórtica, presente en 58 pacientes con SM, 74,3 % del total de casos estudiados.
Otros signos de carácter cutáneo (estrías atróficas), riesgo de neumotórax y ectasia dural(11) no fueron encontrados en la paciente.
Aunque el diagnóstico de la entidad es eminentemente clínico, se describe la realización de estudios genéticos para la identificación de mutaciones en el gen FBN1,(12) lo que no fue posible realizar en este caso, pero cabe señalar que los elementos clínicos fueron suficientes para el diagnóstico.
La paciente fue asistida previamente por otras especialidades, debido a otras afecciones, como fue el caso de oftalmología (operada por luxación bilateral del cristalino, de forma recurrente) y ortopedia (cifoescoliosis); por lo que se sugiere la pesquisa etiológica de estas afecciones por parte de las especialidades correspondientes, como traducción orgánica de una enfermedad sistémica como el SM. De igual manera, la atención primaria de salud como primer eslabón en la atención médica de nuestros pacientes pudo haber identificado signos orientadores de la identidad nosológica. A propósito de lo antes expuesto, en estudio realizado por Figueroa y colaboradores(5) se encontró que la subluxación del cristalino ocurrió en 87 % de los casos con el síndrome, dato que constituyó el principal motivo de consulta y sospecha de SM.
Se describen otras enfermedades con rasgos clínicos similares al SM, por lo que es necesario el diagnóstico diferencial. El síndrome de Marfan-Like es una afección congénita que presenta manifestaciones clínicas similares, pero no cumple con todos sus criterios. El síndrome de Ehlers-Danlos constituye un trastorno del tejido conectivo caracterizado por diversos grados de hiperelasticidad, hipermovilidad y trastornos temporomandibulares. Por otra parte, el síndrome de Shprintzen-Goldberg es un raro trastorno sistémico del tejido conectivo caracterizado por la presencia de manifestaciones marfanoides; un hallazgo distintivo resulta la presencia de discapacidad intelectual, aunque no todos los pacientes lo presentan. Todos estos síndromes muestran una superposición significativa con las características observadas en el SM.(1)
Además, debe diferenciarse del síndrome de hipermovilidad articular benigna, cuyas principales manifestaciones clínicas son la hipermovilidad y el dolor en múltiples articulaciones.(16) Por otra parte, el Síndrome Loeys–Dietz se manifiesta con alteraciones vasculares más agresivas en comparación con SM, asociado a disección aórtica en cerca del 70 % de los casos y una sobrevida que apenas alcanza los 37 años.(17)
Actualmente, el manejo de estos pacientes se realiza teniendo en cuenta medidas de prevención, de rehabilitación, vigilancia y terapéutica oportuna de las complicaciones. El tratamiento con β bloqueadores se recomienda como profiláctico en cualquier paciente con SM y dilatación de la raíz aórtica, ya que puede disminuir su progresión.(12)
Se sugiere no realizar deportes competitivos, de contacto, ejercicios isométricos ni actividades que causen lesión o dolor de las articulaciones. Se debe evitar el consumo de fármacos que estimulen el sistema cardiovascular, como los descongestivos. La cafeína puede agravar la tendencia a las arritmias. También se prohíbe el uso de instrumentos de viento o ventilación con presión positiva por pacientes con tendencia a presentar neumotórax.(12)
Coincidiendo con el criterio de Pérez y colaboradores,(12) es importante la divulgación y estudio de estos casos entre los médicos en formación y profesionales de reciente graduación, pues debido a su baja morbilidad, su diagnóstico puede pasar inadvertido.
El SM es una enfermedad hereditaria que generalmente no se manifiesta en la edad temprana. Los criterios de diagnóstico han sufrido diversas modificaciones desde la descripción inicial de la enfermedad, siendo los más actuales los criterios de Gante revisados, donde para determinar la enfermedad no se requiere de estudios genéticos cuando se reúnen elementos clínicos suficientes. El diagnóstico tardío puede traer consigo la aparición de complicaciones que ponen en peligro la vida del paciente. Se sugiere la pesquisa etiológica de afecciones como luxación del cristalino y escoliosis, por parte de las especialidades correspondientes, como traducción orgánica de una enfermedad sistémica como el SM.
Conflicto de intereses:
Los autores declaran que no existe conflicto de intereses.
Contribución de los autores:
Conceptualización: Laydamí Rodríguez Amador, Idioel Abreu La Rosa, Héctor Javier Cruz de los Santos, Lorena Álvarez Fernández, Ana Laura Taillacq Suárez, Roberto Lloveras Casañas
Investigación: Laydamí Rodríguez Amador, Idioel Abreu La Rosa, Lorena Álvarez Fernández, Ana Laura Taillacq Suárez, Roberto Lloveras Casañas
Metodología: Laydamí Rodríguez Amador, Idioel Abreu La Rosa, Héctor Javier Cruz de los Santos
Redacción – borrador original: Laydamí Rodríguez Amador, Idioel Abreu La Rosa, Héctor Javier Cruz de los Santos, Lorena Álvarez Fernández, Ana Laura Taillacq Suárez, Roberto Lloveras Casañas
Redacción – revisión y edición: Laydamí Rodríguez Amador, Idioel Abreu La Rosa
Financiación:
Autofinanciado.
