INTRODUCCIÓN
La angiomatosis encefalotrigeminal descrita por Sturge(1) en 1879 y redondeada por Weber(2) en 1922, es el más frecuente de los síndromes neurocutáneos con predominio de anomalías vasculares. Es un proceso congénito infrecuente, pero no excepcional, que afecta a uno y otro sexos por igual y que aparece de manera esporádica, aunque se han descrito casos heredados de forma autosómica recesiva y dominante.
El síndrome de Sturge-Weber (SSW) puede ser de forma completa e incompleta. Su forma completa consiste en la asociación de anomalías cerebrales (angioma leptomeníngeo o pial), cutáneas (angioma facial) y oculares (angioma coroideo); desde el punto de vista clínico se caracteriza por una mancha color vino en la cara, epilepsia, retraso mental, otras manifestaciones neurológicas deficitarias(hemiparesia, hemianopsia) y glaucoma. Las formas incompletas del síndrome, aparecen como:( 3)
-Angioma facial y leptomeníngeo, pero sin angioma coroideo.
-Angioma leptomeníngeo y coroideo sin nevus facial.
-Nevus facial y angioma coroideo sin evidencias clínicas ni radiográficas de angiomatosis cerebral.
-Angiomatosis cerebral y pial aislado.
Generalmente, esta angiomatosis leptomeníngea condiciona la presencia de convulsiones focales y de hemiparesia que suelen ser contralaterales al lado afectado, junto con una atrofia cerebral del mismo lado del angioma facial y un retraso mental más o menos marcado. En ocasiones existen también alteraciones oculares por malformaciones vasculares a dicho nivel.(4)
Es un trastorno congénito y esporádico, no hereditario, aunque se han descrito algunos casos familiares.(5) Se ha descrito la asociación de este síndrome con el gen GNAQ.(6) La mutación en la proteína G GNAQ y RASp21 se asocia a SSW y angioma facial no sindromático.(7)
Los factores de peor pronóstico a nivel neurocognitivo son: comienzo de las crisis antes de los seis meses, compromiso leptomeníngeo bilateral y la alta frecuencia de convulsiones. Los episodios de déficit motor (EDM) se manifiestan como hemiparesia transitoria o defectos en el campo visual.(6) Fisiopatológicamente se produce una isquemia transitoria cortical y trombosis recurrentes a nivel de la malformación capilar leptomeníngea.(6)
Se ha propuesto el uso preventivo de aspirina a bajas dosis 3 a 5 mg/kg/día a fin de prevenir o limitar los EDM y deterioro neurológico progresivo.(6,7)
No hay consenso con respecto a la edad oportuna para iniciar la medicación anticonvulsivante, aunque algunos grupos de trabajo la inician con el paciente presintomático, sin convulsiones ni EDM, considerando que es el momento óptimo para intervenir y mejorar el pronóstico motor y cognitivo.(6)
El presente trabajo tiene como objetivo señalar lo oportuno del tratamiento para un mejor pronóstico y calidad de vida.
PRESENTACIÓN DEL CASO
Se presenta el caso de una paciente del sexo femenino, transicional de un año de edad, nacida de un parto eutócico de una madre con historia obstétrica de siete embarazos, cuatro partos y dos abortos provocados, la niña es la quinta de los hijos. No se recogieron antecedentes prenatales de interés, parto eutócico de 39 semanas de gestación, Apgar 8-9, peso 3660 gramos.
Desde el nacimiento presentó un angioma facial extenso. A los tres meses de edad comenzó a padecer cuadros convulsivos con epilepsia focal motora derecha que se generalizó secundariamente, con ingresos reiterados por descompensación de la epilepsia de difícil control, retardo en el desarrollo psicomotor.
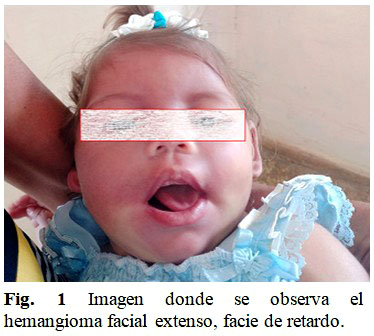
Al examen físico se constató una disminución generalizada del tono, no sostén cefálico, reflejos osteotendinosos abolidos generalizados. Disminución de la motilidad en hemicuerpo derecho (hemiparesia derecha), pulgares ocultos bilaterales, nistagmo bilateral vertical y horizontal. Paresia del VI par craneal izquierdo. No signo de Babinsky, no se sienta, no presenta pinza digital, no gatea. Evidente retraso en el desarrollo psicomotor. En la piel un hemangioma facial extenso bilateral que solo respeta la tercera rama del trigémino derecho. (Fig. 1).
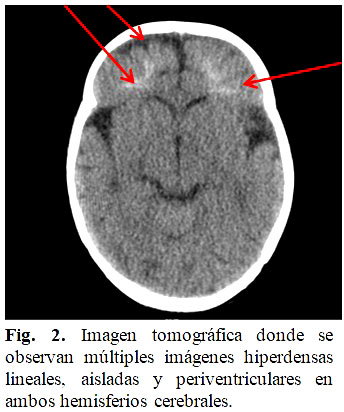
Se le realizó TAC de cráneo, que arrojó: múltiples imágenes hiperdensas lineales, aisladas y periventriculares en ambos hemisferios cerebrales. Atrofia frontal bilateral izquierda mayor que derecha. Impresiona telangestasia capilar. (Fig. 2).
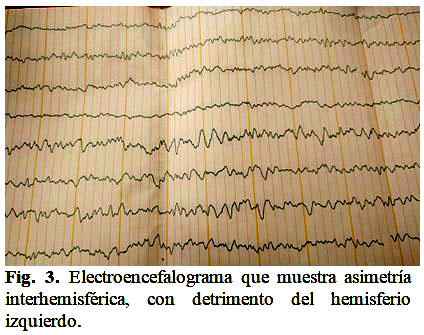
El electroencefalograma del caso arrojó los siguientes resultados: patológico, asimetría interhemisférica, con detrimento del hemisferio izquierdo, signos de irritación cortical parietotemporal derecho. (Fig. 3).
Se comenzó tratamiento con fenobarbital (15mg /5ml) a 5mg/kg/ día, hasta los seis meses de edad, más fenitoína: 5mg/kg/día. Actualmente está con valproato de sodio (250mg/5ml) 40mg/kg/día.
Se insertó en el programa de neurorehabilitación infantil en el centro de Aguada de Pasajeros por ciclos de 28 días observándose mejoría en el desarrollo psicomotor.
Por todos los criterios antes expuestos se realiza el diagnóstico de un síndrome de Sturger Weber.
DISCUSIÓN
El SSW es conocido como completo cuando afecta al sistema nervioso central (SNC) y los angiomas faciales están presentes y se define como incompleto cuando sólo uno está presente; la escala de Roach se utiliza para esta clasificación: (8)
‐Tipo I. Angioma facial y de leptomeninges están presentes; puede tener glaucoma.
‐Tipo II. Angioma facial solamente, no afectado sistema nervioso central ; puede tener glaucoma.
‐ Tipo III. Angioma de leptomeninges solamente; no glaucoma.
El angioma cutáneo es conocido como "mancha en vino de Oporto". Se puede presentar también en angiomas extracraneales y de tejidos blandos. Otras malformaciones del sistema nervioso central se asocian a esta enfermedad y otros síndromes neurocutáneos se incluyen en su diagnóstico diferencial.(9)
El caso presentado tiene un síndrome de Sturger-Weber de tipo I, presenta angioma facial, distribuido en cuero cabelludo y cara, respetando solo la tercera rama del trigémino derecho. En leptomeninges presenta telangectasia capilar, aun no presenta glaucoma.
La obstrucción de las formaciones vasculares en SWS puede causar éstasis en la microcirculación con reducción del retorno venoso, hipoxia y descenso del metabolismo neuronal, siendo el mecanismo patogénico en estos casos. La resolución de la sintomatología y de las alteraciones en la resonancia magnética nuclear (RMN) cerebral sugieren una isquemia reversible, más que un infarto.(10)
El uso preventivo de aspirina a bajas dosis, 3 a 5 mg/kg/ día, sirve para prevenir o limitar los episodios de déficit motor y el deterioro neurológico progresivo. Su uso es controvertido en pediatría, por lo que se recomienda que estos pacientes reciban la vacuna contra la varicela y la anual contra influenza, por la asociación de Síndrome de Reye en pacientes que reciben AAS y estas virosis. (10)
La exploración electroencefalográfica puede ayudar, inicialmente, a confirmar la presencia de afectación cerebral y, más tarde, a localizar el sitio de inicio de las crisis epilépticas. En algunos pacientes con angioma pial unilateral pueden observarse paroxismos generalizados y bisincrónicos. Los estudios de metabolismo cerebral mediante tomografía de emisión de positrones han mostrado una reducción del metabolismo cerebral cerca del angioma que se extiende bien por toda el área radiográfica anormal.(3)
En el caso que se presenta el electroencefalograma localiza el lugar de las crisis en la región parietotemporal derecha, con detrimento del lado izquierdo, en correspondencia con lo planteado por el autor, Morales Querol.(3)
La tomografía de emisión de fotón simple y el estudio de flujo sanguíneo cerebral usando inhalación de xenón demuestran la reducción de la perfusión en el tejido cerebral afectado. Los estudios de imagenología funcional complementan a las estructurales para la evaluación del tratamiento quirúrgico en estos pacientes.(3)
Está discutido el momento oportuno para estudiar estos pacientes con neuroimágenes, teniendo en cuenta los potenciales efectos de deterioro neurocognitivo que produce el uso de sedación y anestesia general. También se ha demostrado el depósito de gadolinio en distintos tejidos del cerebro (ganglios basales, cerebelo) y en médula ósea. El estudio de neuroimagen óptimo es la RMN 3 tesla, con contraste incluyendo T1, T2, Flair, DWI Y SWI. (9)
En el caso presentado solo se realizó tomografía axial computarizada simple, dificultándose realizar la resonancia magnética nuclear.
En cuanto al tratamiento de la epilepsia, es importante la correcta selección de la droga antiepiléptica de acuerdo con el tipo o los tipos de crisis que muestra el paciente y es necesario ser enérgicos por la importancia de las crisis en el pronóstico. En escasos pacientes de corta edad, con afectación unilateral exclusivamente, con epilepsia refractaria y sin retraso mental profundo, está indicado el tratamiento quirúrgico, mediante la resección del área afectada, lobectomía e incluso hemisferectomía. En estos casos es importante asegurar que el otro hemisferio se encuentra totalmente sano, por lo que requiere una exquisita evaluación neuroimagenológica (estructural y funcional) y electroencefalográfica. (8)
El tratamiento de la enfermedad de Sturge Weber es multifactorial y algo polémico. Las drogas de elección suelen ser la fenitoína a dosis de 5-8mg/kg/día dividido (en dos dosis) y el ácido valpróico, a dosis de 20-60mg/kg/día (dividido en dos dosis), en caso de que sean parciales simples o la carbamazepina con 10-20mg/kg/día (dividido en dos tomas) y el ácido valpróico en caso de que sean parciales complejas. En caso de ser generalizadas se puede iniciar con fenitoína o fenobarbital. Es necesario hacer niveles séricos del anticonvulsivante cada 3-6 meses. En caso de refractariedad a una droga se puede agregar una segunda droga antes de pasar a la opción quirúrgica.(11)
La paciente que ahora se presenta ha tenido buena respuesta al tratamiento farmacológico impuesto, consistió al inicio en fenobarbital y fenitoína hasta los seis meses de vida y actualmente con valproato de sodio, coincidiendo con los autores, Samra V y Portillo J.(11)Recibe, además, tratamiento no farmacológico vinculada a la atención temprana donde realiza ciclos de 28 días en neurorehabilitación infantil, con lo que se ha logrado discreta mejoría en su desarrollo psicomotor.
Se presentó un caso representativo de una niña con síndrome de Sturge-Weber, manejo interdisciplinario con los servicios de neurología, oftalmología y pediatría. Se trata de mantener buen manejo interdisciplinario, con el fin de evitar la progresión de dicha lesión y sus consecuencias futuras en el aspecto psicosocial, y mejorar la calidad de vida.
El pronóstico y la calidad de vida dependerán de lo temprano, oportuno y adecuado que sea la vigilancia y el manejo de las convulsiones, la prevención del glaucoma y la ceguera, la estimulación temprana de las áreas motoras, cognitivas y del lenguaje, entrenamiento en destrezas motoras finas, de ser posible todos los días.
Conflicto de intereses
No existen conflictos de intereses.
Contribuciones de los autores
Conceptualización: Marileydis Almaguel García, Irene Estefania Delgado Pérez.
Investigación: Marileydis Almaguel García, José Sánchez Prieto, Irene Estefania Delgado Pérez.
Visualización: Marileydis Almaguel García, José Sánchez Prieto, Irene Estefania Delgado Pérez.
Redacción – borrador original: Marileydis Almaguel García, José SánchezPrieto, Irene Estefania Delgado Pérez.
Redacción, revisión y edición: Marileydis Almaguel García, José Sánchez Prieto, Irene Estefania Delgado Pérez.
Financiamiento
Hospital-Policlínico Miguel Alipio de León Hernández. Aguada de Pasajeros, Cienfuegos. Cuba.
