INTRODUCCIÓN
La polimiositis y dermatomiositis son las formas más frecuentes de miopatías inflamatorias idiopáticas, un grupo heterogéneo de enfermedades adquiridas y sistémicas del tejido conjuntivo que se caracterizan por los efectos clínicos y anatomopatológicos de una inflamación crónica no supurativa del músculo, de causa desconocida.(1,2)
La dermatomiositis tiene su máxima prevalencia en la infancia (7-15 años) y en la mediana edad (30-50 años).(3) La incidencia anual de dermatomiositis es de aproximadamente por 0,8 a 1,2 por 100.000 y la de polimiositis (PM) 0,1 a 3,2 por 100.000 personas.(4) Igualmente significativo es su predominio en el sexo femenino con un aproximado de 3:1 y en ciertos grupos étnicos. Las personas de origen africano o latino tienen mayor riesgo de miopatías inflamatorias idiopáticas y peor pronóstico que los de descendencia europea.(1,5)
En 1916 se señaló por primera vez la asociación entre dermatomiositis y cáncer. En la actualidad parece claro que se asocia a un incremento de la incidencia de neoplasias, siendo los tipos de neoplasias encontradas similares a los detectados en la población en general.(6)
Se caracteriza por progresar con rapidez, tener remisiones comunes y afectación muscular global más que selectiva. El eritema característico que la acompaña facilita su identificación temprana; diferente de lo que ocurre con la polimiositis, cuya fecha real de inicio es difícil de precisar y de manera típica los pacientes retrasan varios meses la consulta con el médico.(5,7)
La afectación visceral más frecuente es el pulmón, con comunicaciones que estiman la prevalencia entre 10 y 60 %. La afección respiratoria más frecuente en pacientes con miopatías inflamatorias es el compromiso intersticial, generalmente de instalación subaguda o crónica.(8)
Por otra parte el neumomediastino espontáneo se ha presentado como entidad clínica de origen desconocido y evolución benigna que afecta a individuos jóvenes.(9) Es una complicación poco frecuente de diversos tipos de enfermedades intersticiales asociadas a patologías del tejido conectivo. La miositis inflamatoria es la enfermedad más frecuente del tejido conectivo asociada con neumomediastino.(10)
En ocasiones se produce una rotura alveolar como resultado de la afección parenquimatosa periférica en enfermedades intersticiales que producen panalización periférica, como es el caso de la dermatomiositis, muchas veces en su forma amiopática. La afección pulmonar en estos casos suele ser especialmente grave, con una elevada mortalidad que solo disminuye si se instaura tratamiento inmunosupresor potente de forma temprana.(8)
La anamnesis médica y familiar cuidadosa, una exploración física completa y las pruebas de laboratorio, sobre todo las enzimas musculares séricas; los exámenes imagenológicos, la electromiografía y la biopsia muscular son claves para el diagnóstico.(3)
Por la baja prevalencia de esta enfermedad y la novedad de su diagnóstico, se decidió la presentación de este caso.
PRESENTACIÓN DEL CASO
Paciente mestizo, masculino, de 47 años de edad y de procedencia rural. Con antecedentes de salud y un ingreso previo tres meses antes por presentar síndrome febril agudo, asociado a un síndrome poliarticular con diagnóstico al egreso de artritis reactiva. Llevó tratamiento sintomático en ese momento.
El día 30 de agosto de 2020, acude al Cuerpo de Guardia del Hospital General Universitario Dr. Gustavo Aldereguía Lima, de Cienfuegos, por marcada pérdida de peso de varias semanas, debilidad muscular proximal discreta, malestar general con eritema en heliotropo y edema facial.
Refirió dolor torácico y disfagia, indistintamente a líquidos y sólidos.
Al examen físico se documentaron como datos positivos:
Piel y mucosas húmedas e hipocoloreadas, con presencia de eritema en heliotropo. Edema de fácil godet en párpados superiores e inferiores. (Figura 1).
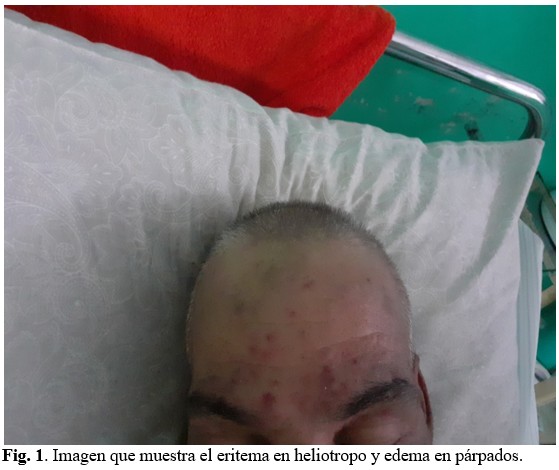
A la auscultación del aparato respiratorio, presencia de estertores crepitantes en base pulmonar derecha.
Refirió debilidad muscular proximal hacia ambos miembros superiores que era muy discreta al momento del ingreso.
En el Cuerpo de Guardia se le realizó radiografía de tórax, en la que se observaron pequeñas opacidades bilaterales de aspecto inflamatorio. En el pulmón derecho, una de las imágenes mostraba tendencia nodular de aproximadamente 1 cm.
Índice cardiotorácico (ICT): normal.
No se contaba con la posibilidad en ese momento de realizar creatinphosphokinasa (CPK) de urgencia, la cual fue indicada teniendo en cuenta la sospecha clínica de una miopatía inflamatoria.
Por lo que se decidió ingreso para estudio y tratamiento en la sala 12 B, de Medicina Interna, con impresión diagnóstica de una dermatomiositis más una neumonía clase II A.
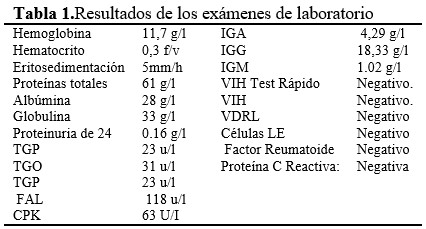
Durante esta etapa de hospitalización se le indicaron los exámenes complementarios indispensables, y otros necesarios para confirmar el diagnóstico y descartar otras afecciones. Entre ellos se pueden destacar: (Tabla 1).
Electrocardiograma (EKG): sin alteraciones.
Ecocardiograma: ventana acústica deficiente, no presencia de trombos, no presencia de derrame pericárdico, no hipertrofia ventricular izquierda (HVI), patrón de relajación normal, no dilatación de cavidades. FSG conservado. FEVI: 70 %
Se decidió comenzar el tratamiento con ceftriaxona (1 gramo) 1 bbo cada 12 horas por 7 días. Teniendo en cuenta la situación epidemiológica generada por la COVID-19, se realizó PCR, que fue negativo.
En sala el paciente presentó una evolución clínica favorable desde el punto de vista respiratorio, pero continuó con los síntomas y signos clínicos sugestivos de miopatía inflamatoria encontrados al ingreso, incluso con empeoramiento de las lesiones en piel, pérdida de peso, atrofia muscular marcada y disfagia.
Se discutió en colectivo con la presencia de las especialidades de Reumatología y Dermatología, se decidió realizar otros estudios para descartar causas secundarias que estuvieran condicionando el cuadro en ese momento y que tuviesen como manifestación la presencia de la dermatomiositis. También se decidió comenzar con el tratamiento empírico con prednisona, 60 mg diarios, teniendo en cuenta el empeoramiento de la disfagia y el dolor torácico. Por esta razón se realizaron los siguientes estudios:
Examen de ORL: sin alteraciones.
Radiografía de ambos hombros: se observan signos de osteoporosis difusos en huesos que conforman ambos hombros. Cambios degenerativos óseos.
Ultrasonido abdominal: hígado que no rebasa el reborde costal, con marcado aumento de su ecogenicidad. No dilatación de vías biliares. Páncreas y bazo de tamaño y ecopatrón normal. Vesícula normal. Ambos riñones de tamaño normal y buen parénquima. Vejiga normal, paredes de tamaño y ecopatrón normal.
Ultrasonido prostático: próstata de contornos bien definidos, ecopatrón homogéneo que mide 38 x 24 x 29 mm.
Endoscopia:
Duodenitis eritematosa moderada.
Úlcera péptica duodenal en fase de cicatrización.
Gastritis eritematosa moderada.
Se indicó tratamiento de triple terapia para el H. Pylori con omeprazol, amoxacilina y claritromicina.
Tomografía axial computarizada de tórax: a nivel de ambos lóbulos inferiores y parcialmente en lóbulo medio derecho se observa engrosamiento de los septos interlobulillares centrales de alta densidad tomográfica e imágenes nodulares subpleurales de mediana densidad. Diagnóstico: fibrosis intersticial.
Colonoscopia (Número 173/20): Normal
Observaciones: Se tomaron muestras de distintos segmentos del colon para su estudio histológico que informó: mucosa cólica donde se observa infiltrado inflamatorio crónico a predominio linfocitario de ligero a moderado, y edema de la lámina propia. Cambios histológicos presentes en todos los segmentos.
A los 11 días de evolución en sala de Medicina Interna se decidió egreso y seguimiento por consulta de Reumatología por mejoría de su cuadro clínico. Pendiente a concluir estudio de su enfermedad. El rayos X de tórax al egreso sin evidencia de lesiones inflamatorias y con persistencia de las lesiones ya diagnosticadas previamente.
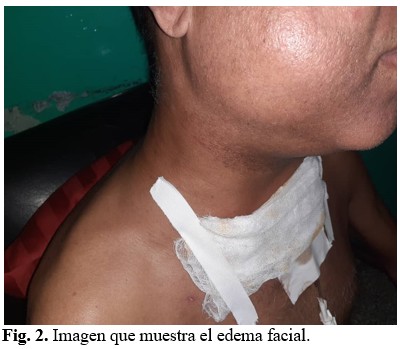
Posteriormente a las 48 horas de su egreso el paciente comienza con aumento de volumen de toda la cara el cuello y región supraclavicular. Se constata al examen físico presencia de enfisema celular subcutáneo. (Figura 2).
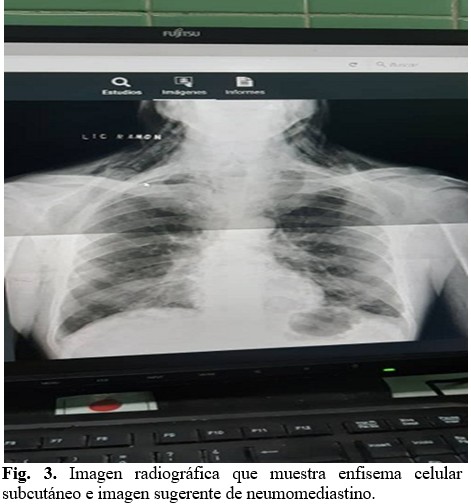
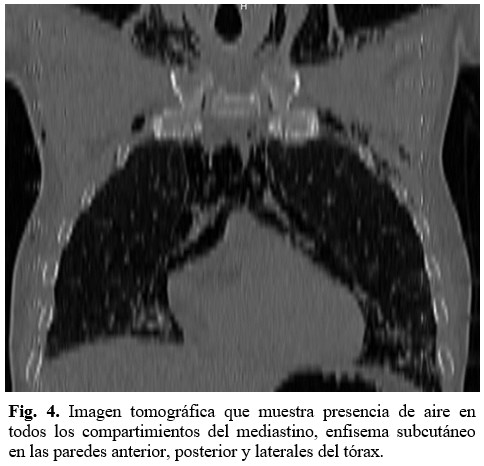
Se realizó rayos X de tórax que informó presencia de enfisema celular subcutáneo e imagen sugerente de neumomediastino (Figura 3), por lo que se indicó estudio tomográfico de tórax que informó presencia de aire en todos los compartimientos del mediastino, enfisema subcutáneo en las paredes anterior, posterior y laterales del tórax. (Figura 4).
Se valoró con la especialidad de Cirugía General, decidiéndose su ingreso en este servicio para tratamiento quirúrgico: mediastinostomía con broncoscopia. Se encontró durante la exploración la presencia de debilidad en la pared posterior de la tráquea, sin otro hallazgo de relevancia y sin complicaciones del acto quirúrgico. Se tomó muestra de suero para estudios inmunológicos cuyos resultados fueron:
ANA negativo
Anticuerpos de miositis:
- Anti-JO-1 negativo
- Anti-Mi2-2 negativo
- Anti-PM-Sd negativo
- Anti-U1-snRNP negativo (inmunobiot)
- Anti-Ku(p70/80) negativo
Factor reumatoideo: 8.4 UL/ml VN:0-14 UL/ml
Se consideró que los resultados negativos estuvieron en relación con el uso de esteroides, que dada su condición fue imposible retirar. Se debe aclarar, además, que no fueron realizados todos los estudios específicos en estos casos por no contar con las posibilidades tecnológicas para hacerlo.
A partir de los hallazgos clínicos y la evolución del paciente hasta el momento, y de la asociación entre la dermatomiositis y los neumomediastino y neumotórax espontáneos, se discutió el caso nuevamente de conjunto con el Servicio de Cirugía General, donde se encontraba ingresado en ese momento el paciente por el tratamiento quirúrgico y se decidió realizar biopsia de piel y músculo.
Biopsia de músculo gastrocnemio izquierdo:
Distrofia muscular focal, acompañado de infiltrado inflamatorio crónico perivascular, constituido por escasos linfocitos y macrófagos más abundantes, hallazgos que pueden corresponder con dermatomiositis que debe ser corroborado por estudios histoquímicos.
Teniendo en cuenta este diagnóstico histológico y el cuadro clínico del paciente se decidió comenzar con tratamiento inmunosupresor con ciclofosfamida 1 gramo mensual y prednisona 40mg diarios, manteniendo el paciente hospitalizado en sala 12B de Medicina Interna y con seguimiento diario por Reumatología y Cirugía General debido a la persistencia del neumomediastino.
DISCUSIÓN
La dermatomiositis es una enfermedad del tejido conectivo de etiología incierta y compromiso multisistémico, que involucra principalmente el sistema músculo esquelético y la piel. Se asocia a complicaciones respiratorias, principalmente la enfermedad pulmonar intersticial (EPI), presente entre el 5 y el 30 % de los pacientes con dermatomiositis.(4)
Existen varias teorías que involucran factores infecciosos, virales, endocrinos, genéticos y autoinmunes en la patogenia de esta entidad. En ocasiones, un porcentaje significativo de pacientes, estimado entre el 15 y 25 %, presentan un proceso neoplásico subyacente. Dentro de los criterios que involucran los factores virales existe vinculación con retrovirus y virus Coxsackie B, los cuales invaden los músculos, dañan el endotelio vascular y causan liberación de citocinas que, a su vez, inducen expresión de genes del complejo mayor de histocompatibilidad que activan una respuesta inmunitaria mediada por células TCD8 citotóxicas que dañan el músculo.(11)
Por otra parte, según las teorías autoinmunes, entre el 60 y el 80 % de los pacientes se les puede detectar autoanticuerpos nucleares y citoplasmáticos específicos o relacionados con miositis (anti-Jo-1, Anti-Mi2-2). Recientemente se han identificado anticuerpos dirigidos contra la proteína codificada por el gen asociado con la diferenciación del melanoma 5 (melanoma differentiation associated gene 5 [MDA5]), perteneciente a la familia de receptores Rig-I-like, que se relacionan con la respuesta a infecciones virales). Se reporta que están presentes en entre el 19 y el 35 % de los pacientes con dermatomiositis clínicamente amiopática y se asocian específicamente con compromiso muscular mínimo o ausente y EPI rápidamente progresiva, complicada, con frecuencia, por la aparición de neumomediastino espontáneo.(9) En el caso que se presenta se muestra un cuadro clínico caracterizado por presentación amiopática, asociado a EPI y neumomediastino espontáneo.
Las manifestaciones clínicas generalmente están relacionadas con la afectación dermatológica y la toma muscular, asociado con manifestaciones generales:(3)
- Debilidad muscular progresiva y simétrica que afecta sobre todo la musculatura proximal de los miembros.
- Debilidad de los músculos faríngeos y flexores del cuello, ocasionando disfagia, presente en nuestro paciente y dificultad para levantar la cabeza (cabeza caída).
- La sensibilidad es normal en esta enfermedad y los reflejos tendinosos están preservados, aunque en los músculos con debilidad o atrofia grave pueden desaparecer.
- Las artralgias, por lo general simétricas, son un síntoma precoz y se observan en el 30 % de los pacientes, presentes también en este caso como pródromo del cuadro actual.
- Las pápulas de Gottron son lesiones elevadas, a menudo descamativas y palpables sobre una base eritematosa en superficies extensoras como las articulaciones metacarpofalángicas e interfalángicas proximales, los codos, las rodillas y son prácticamente patognomónicas.
- El signo de Gottron (máculas en una distribución similar a las pápulas de Gottron).
- Exantema purpúrico, característico en los párpados y a veces rodeándolos llamado exantema en heliotropo.
- Crecimiento excesivo de las cutículas, los cambios periungueales capilares y la dilatación de los capilares en las encías; este último es especialmente frecuente en la dermatomiositis juvenil. Además se pueden encontrar fisuras ásperas, descamativas y eritematosas en las caras palmar y lateral de los dedos, lo que se conoce como manos de mecánico.
- Manifestaciones generales (astenia, pérdida de peso, anorexia, fiebre, fenómeno de Raynaud).
- En la literatura revisada también se describe la presencia de manifestaciones sistémicas donde se incluyen las relacionadas con neoplasias, enfermedad pulmonar intersticial, afecciones del aparato digestivo y cardiopatías.
La presencia de arritmias, insuficiencia cardiaca congestiva, miocarditis y pericarditis son detectables en imágenes y en la electrofisiología, generalmente, en más de 50 % de los casos.(4)
En el caso de las neoplasias las más frecuentes son ovario, mama, colon, melanoma y linfoma no Hodgkin.(4)
La enfermedad pulmonar intersticial se presenta con infiltrados inflamatorios y fibrosis intersticial pulmonar. Aparece entre el 15 y el 30 % de las dermatomiositis, confiriéndoles mayor severidad y peor pronóstico.(4)
Alrededor de 50 % de los pacientes con dermatomiositis tiene anticuerpos anti Jo-1 positivos y pueden presentar lesiones hiperqueratósicas en las manos, fenómeno de Raynaud, fiebre, poliartritis, conformando el síndrome antisintetasa. El compromiso gastrointestinal con dolor abdominal, úlceras y perforación visceral es raro, aparece con más frecuencia en las formas juveniles reportadas. La dermatomiositis y la polimiositis pueden estar asociadas con otras conectivopatías dando lugar a los síndromes de sobreposición.(4)
En el caso que se presenta la enfermedad no ha evolucionado según las formas clásicas descritas. No ha predominado la toma de los músculos proximales, pero han estado presentes la disfagia, las lesiones dermatológicas (exantema en heliotropo), las manifestaciones generales y sistémicas, acompañadas de EPI.
Los exámenes complementarios para realizar el diagnóstico final de la enfermedad transitan desde exámenes de analítica sanguínea, inmunológicos y biopsia de piel y músculo.
Creatinquinasa (CK): es sensible, pero no se correlaciona con la severidad de los síntomas y varía con el tratamiento. En la dermatomiositis la CK se eleva, en el 80-90 % de los casos, hasta 50 veces el límite superior normal. El compromiso perimisial selectivo, puede generar un aumento de la aldolasa sin elevación de CK, describiéndose en el síndrome antisintetasa.(4)
Otros exámenes: otras enzimas musculares son menos sensibles. La velocidad de eritrosedimentación generalmente es normal, pudiendo elevarse cuando hay neoplasias y otras conectivopatías.(4)
Anticuerpos asociados a miositis: los más frecuentes son los anticuerpos anti RO52, encontrándose en 19 a 25 % de las miopatías inflamatorias inmunológicas asociándose a EPI.(4)
Anticuerpos específicos de miositis: los anticuerpos antisintetasas, los más frecuentes, aparecen en 35 a 40 % de los pacientes. De estos, los anticuerpos anti Jo1, son los más frecuentes, se ven en más de un 20 %. El resto de los anticuerpos antisintetasas aparecen en menos de 5 %. Los anti Mi-2 se encuentran entre 10 a 30 % de las miopatías, predominantemente en la dermatomiositis, asociándose a respuesta favorable a corticoides y mejor pronóstico.(4)
Tamizaje de neoplasias: el riesgo de neoplasias ocurre en todas las miopatías inflamatorias idiopáticas, siendo mayor en la dermatomiositis.(3) El 25 % de los adultos con dermatomiositis y 10 % de los pacientes con polimiositis presentan una neoplasia. Tienen un riesgo alto hombres, mayores de 45 años, las personas con enfermedad muscular y/o cutánea severa y marcadores inflamatorios elevados. El mayor riesgo ocurre 2 años antes y 3 años después del diagnóstico.(4) La Federación Europea de Sociedades Neurológicas recomienda un seguimiento con tomografía axial computarizada de tórax, abdomen y pelvis. Agregándose ecografía pélvica y mamografía en mujeres, ecografía testicular en hombres y colonoscopia en mayores de 50 años. Si el tamizaje inicial es negativo, deben repetirse los exámenes luego de 3-6 meses, posteriormente cada 6 meses por cuatro años. Determinación periódica de marcadores tumorales séricos puede agregarse.(4)
Anatomía patológica: la atrofia muscular perifascicular es distintiva, sin embargo puede estar ausente hasta en 50 % de los casos según algunos autores. Además, hay infiltrados inflamatorios perivasculares y perimisiales, preferentemente linfocitos CD4+.4 Muchos de estos cambios antes descritos fueron apreciados en la biopsia muscular del paciente.
Diferentes criterios se han propuesto para el diagnóstico de las miopatías inflamatorias idiopáticas. Bohan y Peter(3) propusieron para el diagnóstico de la dermatomiositis uno de los primeros y más utilizados, que incluye:
1. Debilidad simétrica de musculatura de cinturas y de flexores anteriores del cuello que progresa en semanas a meses.
2. Enzimas musculares elevadas especialmente la CK y a menudo la aldolasa.
3. EMG miopática con potenciales unidad motora pequeños, corta duración y polifásicos. Aumento actividad insersional, fibrilaciones y ondas agudas positivas.
4. Biopsia muscular anormal: degeneración, regeneración, necrosis e infiltrados mononucleares intersticiales.
5. Rash típico (Pápulas de Gottron, signo de Gottron y eritema heliotropo).
Para llegar al diagnóstico de dermatomiositis se otorga un punto a cada criterio y se ponderan los criterios del 1 al 4.
Diagnóstico
1. Definitivo: punto 5 + 3 puntos del 1 al 4
2. Probable: punto 5 + 2 puntos del 1 al 4
3. Posible: punto 5 + 1 puntos del 1 al 4
En el caso que se presenta se puede plantear una enfermedad probable, ya que el único criterio que no se evidenció fue la tríada electromiográfica.
El tratamiento de la dermatomiositis busca mejorar la potencia muscular y con ello también la función de los músculos en actividades de la vida diaria además de reducir las manifestaciones extramusculares (disfagia, disnea, fiebre). Los fármacos utilizados en el tratamiento son: glucocorticoides (prednisona) e inmunodepresores (azatioprina, metotrexato y la ciclofosfamida).(12) Más de la mitad de los pacientes presentan remisiones y exacerbaciones múltiples a lo largo de la enfermedad. Factores implicados en un peor pronóstico y menor supervivencia son la edad avanzada, el retraso en iniciar la terapia con corticoides, la disfagia faríngea con neumonía por aspiración, la neoplasia, la EPI y la afectación miocárdica.
La discapacidad también se relaciona con las complicaciones del tratamiento con corticoides como pueden ser la necrosis ósea avascular y los aplastamientos vertebrales. Otras causas de morbimortalidad secundarias al tratamiento son las infecciones oportunistas, diabetes mellitus, cataratas, hipertensión y la úlcera péptica.(13)
Se puede concluir que la dermatomiositis es una enfermedad con una baja prevalencia y etiología que no está totalmente esclarecida. En este paciente la evolución clínica y las pruebas complementarias permitieron definir el caso como una dermatomiositis, asociada con una complicación de la enfermedad que fue la presencia de enfermedad pulmonar intersticial, con neumomediastino espontáneo como forma de presentación de la misma e iniciar el tratamiento sistémico adecuado. El paciente se mantiene en evolución en estos momentos.
Conflicto de intereses
Los autores plantean que no poseen conflicto de intereses.
Contribuciones de los autores
Conceptualización: Teresa Fonseca Fernández, Yanelka Bouza Jiménez.
Visualización:Yanet Rodríguez Zulueta.
Redacción: Teresa Fonseca Fernández; Alejandro Muñoz Morales.
Redacción, revisión y edición: Teresa Fonseca Fernández, Yanelka Bouza Jiménez.
Financiación
Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. Cuba.
