INTRODUCCIÓN
Los síndromes de osteòlisis idiopática representan un grupo heterogéneo de enfermedades caracterizadas por destrucción y reabsorción de huesos afectados, con aparición de lesiones osteolíticas. Estos síndromes difieren en la presencia o ausencia de transmisión genética, las características clínicas asociadas y la localización principal de la osteòlisis.(1)
Dentro de la clasificación de los síndromes de osteòlisis idiopática, la enfermedad de Gorham-Stout (síndrome de Gorham), ocupa el cuarto lugar. También es conocida como enfermedad del hueso evanescente, huesos fantasmas, o de los huesos perdidos. Es un cuadro clínico caracterizado por la destrucción progresiva de tejido óseo (osteòlisis) y proliferación vascular, con angiomatosis y linfangiomatosis ósea, que produce una osteòlisis progresiva del esqueleto, con pérdida de masa ósea en las áreas afectadas.(1,2)
Por ser un síndrome extremadamente raro y para mostrar la utilidad de los estudios imagenológicos en su diagnóstico se decidió la publicación de este caso.
PRESENTACIÓN DEL CASO
Paciente femenina, de dos años de edad, nacida producto de embarazo de alto riesgo obstétrico por madre adolescente, parto pretérmino a las 34 semanas, con un peso al nacer de 2000 gramos. Se recogió como antecedente haber padecido de íctero fisiológico agravado. Requirió de dos ingresos en el primer año de vida por síndrome febril inespecífico. En esta ocasión acudió al hospital por presentar cuadro febril de 38 grados Celsius de una semana de evolución, acompañado de aumento de volumen de la región dorsal. Los familiares refirieron, además, una pérdida de peso de aproximadamente tres kilogramos.
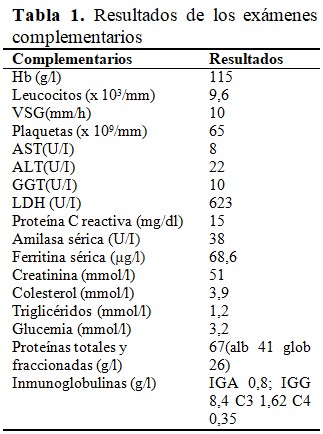
Al examen físico se constataron como datos positivos palidez cutáneo-mucosa e hiperlaxitud articular generalizada, además de una lesión nodular indolora de aproximadamente tres centímetros, eritematosa, en la región dorsal, y otra similar en la cara lateral de la rodilla derecha. No obstante, su estado general impresionaba como una niña no severamente enferma. Se decidió su admisión en sala de Pediatría General y se indicaron exámenes complementarios, que arrojaron los siguientes resultados: (Tabla 1).
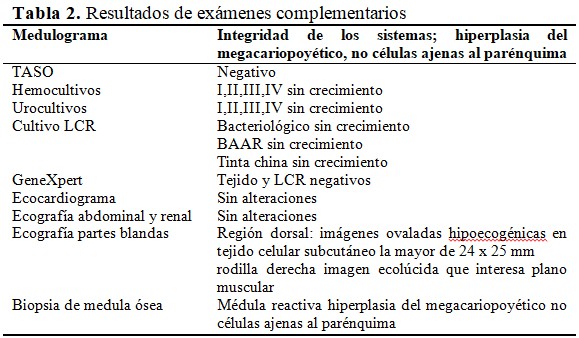
Se recogió el antecedente familiar del padre haber sido contacto en la niñez de un adulto con tuberculosis BK + que llevó tratamiento quimioprofiláctico. Se realizó prueba de tuberculina a la paciente y a todos sus contactos directos de primera línea y la lectura fue de 0 mm en todos los casos. Durante los días sucesivos la fiebre remitió y comenzó con manifestaciones catarrales. A la semana de hospitalización reapareció la fiebre y se observó un marcado aumento de volumen en la región dorso lumbar. La paciente impresionaba agudamente enferma con facie dolorosa y con limitación marcada de su movilidad en el lecho y llanto intenso a las maniobras de exploración. Se detectaron áreas equimóticas en las extremidades y tronco. Se realizó coagulograma mínimo que mostró cifras de trombocitopenia severa (5x109). Se decidió su traslado a sala de Cuidados Intermedios. Fue reevaluada, se indicaron nuevos complementarios específicos. (Tabla 2).
Se decidió iniciar tratamiento antibiótico con ceftriaxone y dada la severa trombocitopenia se inició esquema con gammaglobulina endovenosa a razón de 400 mg/Kg/día en cinco dosis. La fiebre desapareció y se restablecieron los valores normales de plaquetas.
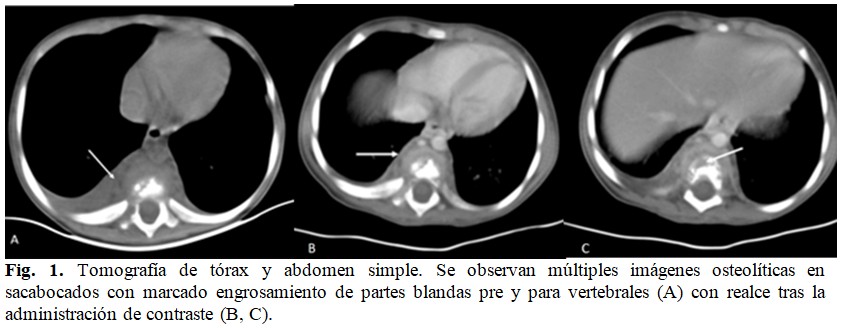
Se realizó una tomografía axial computarizada (TAC) de tórax y abdomen que informó la presencia de múltiples imágenes líticas, que se extienden desde D9 a L4, en sacabocados, con mayor destrucción de cuerpos vertebrales superiores respetando lámina, procesos transversos y los discos intervertebrales. Dicho proceso se asociaba a marcado engrosamiento de las partes blandas anteriores a este plano óseo. (Fig. 1).
Dada la estabilidad clínica se realizó una biopsia por incisión de la región lumbar que informó la presencia de fragmentos de tejido conectivo con áreas vasculares, áreas de hemorragia, proliferación de células endoteliales con pequeñas áreas de hematíes en su interior.
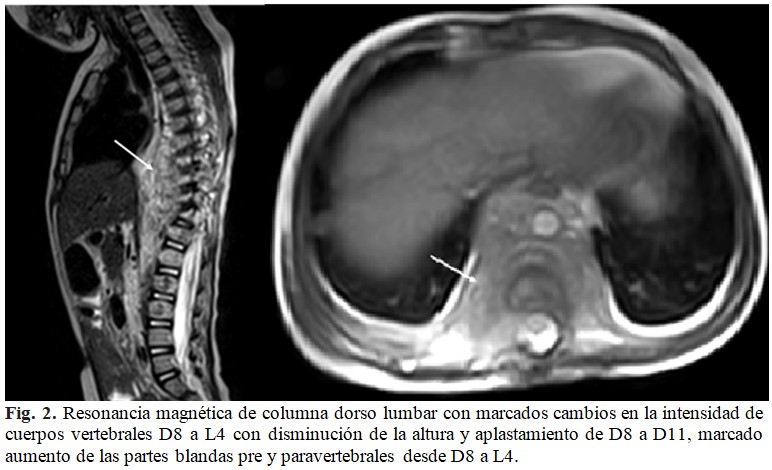
En días sucesivos se mantuvo sin fiebre, ni signos de trombocitopenia, pero con un marcado aumento de volumen de la región dorsal. Se solicitó nuevo estudio tomográfico contrastado que demostró realce del tejido adyacente a los cuerpos vertebrales. Se indicó, además, gammagrafía ósea que describió focos de hipercaptación de radiofármaco de moderada intensidad desde D5 a D8 y L1. Con el objetivo de valorar con mayor resolución estructuras raquimedulares se solicitó resonancia magnética (RM) dorso lumbar que informó marcados cambios en la intensidad de cuerpos vertebrales desde D8 a L4, que se comportaba hipointenso en T1 e hiperintenso en T2 y STIR, disminución de la altura con aplastamiento desde D8 a D11. Se acompañaba de aumento del grosor y cambios en la intensidad de señal de las partes blandas pre y paravertebrales desde D8 a L4. Llamó la atención que las lesiones respetaban los discos vertebrales manteniendo todos la altura e intensidad normal. (Fig. 2).
Se rediscutió en comisión multidisciplinaria y se realizó nueva biopsia transpeduncular obteniendo fragmentos de tejido prevertebral que mostraron una compleja proliferación vascular, con estudios de marcadores inmunohistoquímicos CD34 y alfa actina positivos, cromogranina e inhibina negativos, por tanto se concluyò como una angiomatosis ósea múltiple.
Tras el diagnóstico, la paciente fue evaluada por un equipo multidisciplinario para decidir la estrategia terapéutica. Por la localización y extensión de las lesiones, el tratamiento quirúrgico y la radioterapia fueron descartados. Inició tratamiento con interferón α 2b y propanolol, sin lograr una reducción de las lesiones, sino que, muy por el contrario, se produjo un aumento de la misma hasta generar una marcada deformidad de la región dorsal (cifosis), y manteniendo niveles de trombocitopenia severas que le provocaron una hemorragia intracraneal extensa que causó la muerte a los 18 meses del diagnóstico confirmatorio.
DISCUSIÓN
Las manifestaciones clínicas de la paciente al ingreso, dadas por síntomas generales y tumefacción en la región dorsal, obligaron a descartar en primer lugar etiologías infecciosas y neoformativas.
El antecedente de padre con historia previa de exposición a la tuberculosis hizo plantear en primer lugar la posibilidad de una tuberculosis ósea (mal de Pott). Este planteamiento fue descartado por los resultados de los estudios moleculares microbiológicos (Gene-Xpert) para Mycobacterium tuberculosis. Además, los hallazgos en los estudios anatomopatológicos mostraron ausencia de lesiones granulomatosas calcificadas y ausencia de compromiso del disco intervertebral (discitis) en los exámenes imagenológicos practicados (TAC y RM) de columna vertebral, a pesar de las severas alteraciones de los cuerpos vertebrales con destrucción por aplastamiento. Otras causas infecciosas comunes de tumefacción dorsal como los abscesos paraespinales, generalmente producidos por Staphylococcus aureus, y las osteomielitis vertebrales fueron también descartados por los resultados negativos de los estudios microbiológicos de la lesión, así como de hemocultivos de sangre periférica, y exámenes serológicos para microrganismos como Brucellas.
Las afecciones neoformativas malignas cobraron un peso importante en la discusión sobre todo por la rápida y marcada afectación de los tejidos blandos y la presencia concomitante de síntomas generales (fiebre, pérdida de peso, anorexia), acompañada por manifestaciones purpúricas y trombocitopenia severa en los exámenes complementarios. Se valoró, en primer lugar, teniendo en cuenta la edad de la paciente, la posibilidad de un neuroblastoma metastásico, pero los resultados histológicos negativos de la lesión, además de la ausencia de células ajenas al parénquima medular en el medulograma realizado lo descartaron. Otras posibilidades etiológicas neoplásicas como los sarcomas de musculo o hueso, fueron también descartadas por los resultados histopatológicos y los marcadores de inmunohistoquímica negativos.
La trombocitopenia severa se manejó en principio como de posible etiología inmunológica, pero la ausencia de anticuerpos antiplaquetarios, y la falta de respuesta al tratamiento con intacglobin orientó a una destrucción periférica por el secuestro a nivel del tejido vascular (angiomatosis con consumo de plaquetas y fibrinógeno) debido a un síndrome de Kasabach Merritt.
El planteamiento definitivo de síndrome de Gorham-Stout se llegó al cumplir con todos los criterios requeridos:( 3,4)
- Evidencia de proliferación vascular (patrón angiomatoso, incluye vasos arteriales, venosos, o sistema linfático).
- Ausencia completa de atipia celular en las biopsias realizadas.
- Ausencia de calcificaciones distróficas.
- Evidencia de reabsorción progresiva del hueso nativo.
- Ausencia de evidencia de lesiones ulcerativas, o expansivas.
- Ausencia de compromiso visceral.
- Actividad osteolítica demostrada por estudios de imagen.
- Ausencia de evidencia diagnóstica, después de la realización de exámenes complementarios, de etiología infecciosa, inmunológica, metabólica, y hereditaria.
El síndrome (enfermedad) de Gorham-Stout es una enfermedad rara en la que se produce una reabsorción espontánea y progresiva del hueso sin conocerse todavía su mecanismo patogénico. El primer caso fue descrito en 1838 por Jackson, y en 1955 Gorham y Stout definieron específicamente la enfermedad e hicieron una revisión bibliográfica de 24 casos. Hasta la fecha son cerca de 300 el número de casos publicados en la literatura por lo que el síndrome de Gorham-Stout es extremadamente raro. En Cuba se reporta solo un caso en el año 1999.(5)
Este síndrome se denomina de muy diferentes formas, tales como osteòlisis masiva, enfermedad de los huesos evanescentes, absorción espontánea del hueso o hueso fantasma, pero haciendo referencia siempre a una misma entidad.(6,7,8,9) Se caracteriza por osteòlisis, proliferación linfática y angiomatosis sin regeneración ósea, que condiciona limitación funcional y deformidades. La ausencia de datos clínicos específicos hace de su diagnóstico un desafío que finalmente se logra por alta sospecha y exclusión de otras patologías, lo que no ha permitido estandarizar el tratamiento.(9,10,11)
Los sitios más afectados son vértebras, pelvis, costillas y escápulas; en las extremidades superiores el hueso más afectado es el húmero, y en las inferiores, el fémur y la tibia. Sin tener una predilección de sexo, se presenta mayormente en niños y adultos jóvenes.(3,4,9)
Por su falta de predictibilidad, el pronóstico varía mucho en cada paciente, obedece al área afectada y la estabilización o no de la enfermedad. Esta, generalmente, se autolimita y sus complicaciones dependen del lugar y la extensión de su presentación, pero puede llegar a ser fatal como en el caso que se presenta. La afección de las extremidades inferiores o de la cintura pélvica es de buen pronóstico, la afectación de la columna cervical se asocia a una mortalidad del 33 %, que asciende al 52 % cuando cursa con quilotórax secundario a la toma pleural. En la forma espinal de la enfermedad de Gorham Stout se reporta que el 46 % de los casos ocurre a nivel de las vértebras torácicas.(8,9,10,11)
En relación con el tratamiento de la enfermedad se han ensayado diferentes modalidades que van desde técnicas de resección quirúrgica, reconstrucción usando injertos óseos y/o prótesis quirúrgica, según la extensión y localización de la lesión vascular, tratamientos con radioterapia, y opciones farmacológicas. Estas últimas han incluido octreotide, calcitonina, bevacizumab (anticuerpo monoclonal anti‑VEGF‑A), propranolol, interferon alfa-2b, sirolimus, heparina de bajo molecular y bifosfonatos, sobre todo de tercera generación. Estos fármacos se han utilizado de forma independiente o combinada con el objetivo de disminuir la proliferación celular vascular (angiogénesis), y de inhibir la actividad osteoclástica. Si bien han sido utilizadas todas estas opciones, no existe un tratamiento definitivo y aún muchos de ellos están bajo estudio. La rareza de la enfermedad hace que las mayores series sean con muy escasos reportes y prácticamente testimoniales.(7,8,9,10,11,12) En algunos casos se ha reportado una regresión espontanea.(13)
La enfermedad de Gorham es una rara y peculiar condición patológica mùsculo-esquelética en la cual el hueso virtualmente se desintegra y es reemplazado por tejido conectivo vascular. Su etiología es especulativa, con una presentación clínica muy variable. Los estudios imagenológicos resultan de gran utilidad. Posee un pronóstico indeterminado a pesar de las opciones terapéuticas empleadas.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Contribuciones de los autores
Conceptualización: Ariel Moya Machado.
Investigación: Ariel Moya Machado, Leidelén Esquivel Sosa, Yisel González-Ríos.
Visualización: Ariel Moya Machado, Leidelén Esquivel Sosa, Yisel González-Ríos.
Redacción - borrador original: Ariel Moya Machado .
Redacción – revisión y edición: Ariel Moya Machado, Leidelén Esquivel Sosa, Yisel González-Ríos.
Financiación
Hospital Provincial Pediátrico Universitario José Luis Miranda. Villa Clara. Cuba.
