INTRODUCCIÓN
Las enfermedades raras, en la mayor parte de los casos, tienen origen genético, suelen iniciar en la infancia y se estima que existen alrededor de 8000. La mayoría se manifiesta en la edad pediátrica y comparten elementos comunes como el diagnóstico tardío, gravedad, cronicidad, carácter incapacitante, ausencia de tratamiento efectivo, así como el desconocimiento de los profesionales sanitarios y de la sociedad en general.(1)
La paquioniquia congénita (PC) pertenece a este grupo de enfermedades raras, es una genodermatosis poco frecuente de tipo displasia ectodérmica, cuyo rasgo característico es la hipertrofia y distrofia ungueal.(2) Fue descrita por primera vez por Muller en 1904 y luego por Wilson en 1905 y la asociación con queratodermia palmo-plantar y otros defectos ectodérmicos fue reportada por Jadassohn y Lewandowsky en 1906.(3)
Su transmisión ocurre principalmente de forma autosómica dominante, con expresividad variable y penetrancia completa. Se presenta por igual en ambos sexos y clínicamente puede manifestarse desde el nacimiento.(2) La clasificación clínica clásica incluye dos subtipos: subtipo 1 (PC-1 o síndrome Jadassohn-Lewandowsky) y subtipo 2 (PC-2 o síndrome Jackson-Lawler).(4)
El objetivo de este reporte de caso es describir las manifestaciones clínicas, el diagnóstico y tratamiento del primer caso en Cuba con edad pediátrica portador de esta infrecuente entidad.
PRESENTACIÓN DEL CASO
Escolar masculino, de siete años de edad, con antecedentes de uñas amarillas e hipertróficas desde los nueve meses. Al interrogatorio se obtuvo como antecedente patológico familiar que su padre padecía de paquioniquia congénita.
Fue hospitalizado en el Servicio de Clínicas Pediátricas con diagnóstico inicial de escabiosis infectada por tercera ocasión en el plazo de dos meses, sin evolución favorable a la terapéutica empleada.
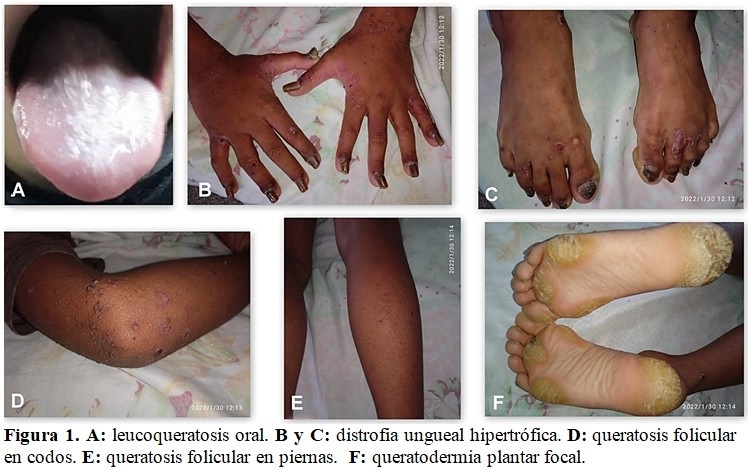
Al examen físico de la mucosa oral, la lengua estaba engrosada, con placas leucoqueratósicas. (Fig. 1.A).
Se observó marcado engrosamiento de todas las uñas de manos y pies, con intensa hiperqueratosis de coloración pardo amarillenta, aumento de las estrías longitudinales y elevación de la uña en su borde libre. (Fig. 1.B y Fig.1.C).
En la piel se constató la presencia de pequeñas pápulas hiperqueratósicas perifoliculares diseminadas, con predominio en la articulación de los codos, manos y miembros inferiores que no confluían, no pruriginosas, algunas de ellas mostraban signos de infección local que fueron refractarias al tratamiento con cremas antibióticas. (Figura 1.D y Fig.1.E).
Presentaba dolor al deambular y con el uso de calzado, debido a la presencia de callosidades de color amarillo, de marcado relieve, forma irregular, duras, gruesas, con fisuras lineales y fondo seco, profundas, localizadas en todo el tercio anterior y posterior de la zona plantar, que respetaban la porción media de ambos pies. (Fig. 1.F).
No se reportaron antecedentes de dientes natales, ni se constataron lesiones quísticas en piel (esteatocitomas). Resto del examen físico sin alteraciones.
Se solicitó interconsulta con los servicios de Dermatología y Genética, al considerar que las lesiones en la región del dorso de las manos, codos y miembros inferiores, inicialmente interpretadas como escabiosis, constituían realmente una queratosis folicular.
Fueron realizados estudios de laboratorio clínico que informaron leucocitosis ligera con predominio de polimorfonucleares y eritrosedimentación acelerada, en relación con la piodermitis secundaria. Se tomó cultivo bacteriológico de las uñas que resultó negativo, al igual que el examen de KOH, por lo cual se descartó la infección micótica. Se obtuvo muestra de la piel de los codos para biopsia que reportó hiperqueratosis. No se confirmó el diagnóstico molecular por no disponer en el país.
Al tener en cuenta las características clínicas descritas en el paciente: la leucoqueratosis oral, la distrofia ungueal hipertrófica, la queratosis folicular y la queratodermia plantar focal; así como la ausencia de esteatocistomas múltiples y dientes natales, sumados al antecedente patológico familiar que evidenció un patrón de herencia autosómico dominante, se consideró como diagnóstico compatible el síndrome Jadassohn-Lewandowsky o PC-1.
Actualmente, el niño mantiene seguimiento por consulta con un equipo multidisciplinario, no han aparecido infecciones secundarias, muestra buen estado psicológico y se integra de manera adecuada con sus congéneres.
DISCUSIÓN
El Registro Internacional de Investigación de Paquioniquia Congénita (IPCRR, del inglés, International PC Research Registry) ha identificado a nivel mundial hasta enero de 2020, 977 individuos en 517 familias confirmadas genéticamente.(3) Su prevalencia se ha estimado en 0,9 por 1 000 000 de individuos.(5) En una amplia búsqueda especializada, no existen reportes publicados de casos clínicos en la edad pediátrica en Cuba.
En la actualidad, la clasificación se realiza en función del gen mutado implicado en la síntesis de queratina: PC-K6a (causada por mutaciones en KRT6A, identificadas en el 44 % de las familias), PC-K6b (KRT6B, en el 5 % de las familias), PC-K6c (KRT6C, en el 2 % de las familias) localizados en 12q13.13, PC-K16 (KRT16, en el 25 % de las familias) y PC-K17 (KRT17, en el 24 % restante) en 17q21.21,3. Las mutaciones localizadas en KRT6A y KRT16 corresponderían clínicamente con PC-1; mientras las localizadas en KRT6B y KRT17 corresponden con PC-2.(6)
Existe cierta correlación fenotipo genotipo en los 2 subtipos. El PC-1 presenta con más frecuencia leucoqueratosis oral (88 % en mutaciones deKRT6A, 41 % en KRT16), mientras los dientes natales y esteatocitomas son más característicos de PC-2 (76 y 67 %, respectivamente en KRT17).(6)
El paciente que nos ocupa presenta las características clínicas de la PC-1, para la cual se han descrito cuatro signos clínicos principales: la distrofia ungueal hipertrófica, queratodermia palmo-plantar dolorosa, queratosis folicular y por último, leucoqueratosis oral.
La distrofia ungueal hipertrófica se caracteriza por una curvatura acentuada con engrosamiento amarillo-marronáceo de los dos tercios distales de las uñas y de los restos subungueales responsables de la inclinación de estas, además de la finalización prematura de la lámina ungueal. La queratodermia palmo-plantar dolorosa puede ser localizada, distribuirse predominantemente sobre las zonas de mayor presión, o ser generalizada y presentarse sobre toda la superficie plantar; junto con hiperhidrosis palmo-plantar, fisuras en zonas de mayor fricción y aparición ocasional de ampollas e infecciones secundarias. La queratosis folicular localizada, sobre todo a nivel del tronco, los glúteos y las áreas extensoras de las extremidades, puede confluir formando placas engrosadas de aspecto verrucoso. Por su parte, en la leucoqueratosis oral no se ha reportado potencial maligno.(7, 8)
El diagnóstico de la PC se basa en hallazgos clínicos; sin embargo, la confirmación genética es imprescindible. La distrofia de las uñas es la característica clínica más precoz, además de ser la que se objetivará con mayor frecuencia en los pacientes pediátricos, presente desde el nacimiento en casi el 40 % de los recién nacidos afectados.(8)
En este escolar no se pudo clasificar la enfermedad según la mutación específica, ya que no se dispone en Cuba del estudio molecular necesario para su diagnóstico.
El tratamiento de la PC es muy complejo y representa un reto para el clínico pediatra. Está orientado a contrarrestar el exceso de queratina de las uñas, piel y mucosas, así como prevenir la aparición de quistes de queratina en la epidermis, de ampollas y del dolor asociado.(2,3)
Para las uñas se recomienda tratamiento oclusivo con urea al 20-40 % y el ácido salicílico al 15-20 %. En otras publicaciones se reporta el uso exitoso de retinoides e isotetrinoína.(2)
Sin embargo, aún no existe un tratamiento que sea específico en la PC. Cuando el engrosamiento ungueal no mejora con el tratamiento tópico, se deberá de realizar la extracción quirúrgica extensa de la lámina ungueal, el lecho y la matriz, seguidos de un injerto cutáneo.(8, 9) Asimismo se han reportado algunos casos tratados de forma exitosa con toxina botulínica.(10,11)
En este paciente se prescribieron emolientes y agentes queratolíticos con un buen control de la enfermedad. Actualmente mantiene seguimiento por consulta con un equipo multidisciplinario, no han aparecido infecciones secundarias, muestra buen estado psicológico y se integra de manera adecuada con sus congéneres.
Es importante la aplicación de medidas conservadoras como reducir la fricción, evitar el trauma y mantener la hidratación adecuada de la epidermis, así como para la mejoría del dolor y la apariencia, relevantes en su vida social. Eventualmente, será importante que el paciente y su familia tengan la oportunidad de acceder a estudios genéticos que permitan identificar la variante patogénica del gen mutado y recibir consejo genético para esta rara enfermedad.
Se concluye que el caso presentado es el primer reporte en Cuba de síndrome de Jadassohn-Lewandowsky en la edad pediátrica, al tener en cuenta las características fenotípicas y los antecedentes familiares.
Conflicto de intereses
Los autores no declaran conflicto de intereses.
Contribuciones de los autores
Conceptualización: Migdalis Hidalgo Muñiz, Aracelis Hernández García, Andrés Andrés Matos, Yadelis Maldonado Martínez.
Visualización: Migdalis Hidalgo Muñiz, Aracelis Hernández García, Andrés Andrés Matos, Yadelis Maldonado Martínez.
Redacción- borrador original: Migdalis Hidalgo Muñiz, Aracelis Hernández García, Andrés Andrés Matos.
Redacción–revisión y edición: Migdalis Hidalgo Muñiz, Aracelis Hernández García, Andrés Andrés Matos, Yadelis Maldonado Martínez.
Financiación
Hospital Pediátrico Provincial Octavio de la Concepción de la Pedraja. Holguín. Cuba.
