INTRODUCCIÓN
La enfermedad de la orina con olor a jarabe de arce (EOJA), leucinosis o cetoaciduria es un error innato del metabolismo, causada por deficiencia en la actividad de la deshidrogenasa de los cetoácidos de la cadena ramificada. Fue descrita por primera vez por Menkes y sus colaboradores en 1954, en cuatro pacientes que presentaron un grave deterioro neurológico en los primeros siete días de vida en los que su orina tenía un olor característico (similar a los extractos de hojas de arce) y que finalmente fallecieron. Presenta una frecuencia media en todo el mundo de 1/185.000 recién nacidos, aunque existen grupos étnicos con incidencia tan alta como 1 en 200 nacimientos. Es más frecuente en las poblaciones con alta tasa de consanguinidad, como en los Menonitas y Oriente Medio.(1,2,3,4,5) En Cuba solo se reporta un caso publicado en la provincia de Matanzas.(6)
Las consecuencias de la EOJA generan una cascada de eventos fisiopatológicos que afecta principalmente el sistema nervioso central lo cual explica la necrosis a nivel de los ganglios basales, aunque no se encuentra dentro de las principales causas de hipodensidad bilateral en tomografía computarizada a nivel.(4)
La necrosis de los ganglios basales es una entidad poco frecuente en la edad pediátrica, puede ser de causa idiopática y como consecuencia de diversas entidades nosológicas como encefalopatías, acidemias, intoxicaciones, traumatismos, entre otros.(1)
Se manifiesta clínicamente por procesos agudos o crónicos, alteraciones piramidales, extrapiramidales, deterioro neurológico, convulsiones o alteraciones del comportamiento.(2,3)
En la mayoría de los casos no se ha encontrado una importante hipotensión o hipoxia previa a la lesión, sino una severa acidosis metabólica.(1)
El objetivo de este trabajo consiste en presentar un caso de enfermedad de la orina con olor a jarabe de arce que en estudio tomográfico mostró hipodensidad de los ganglios basales por necrosis en espejo, entidad muy infrecuente.
PRESENTACIÓN DEL CASO
Escolar de 9 años de edad, con antecedentes de ser producto de embarazo con bajo riesgo obstétrico, parto sin complicaciones y adecuado peso al nacer.
Poseía antecedentes de un ingreso hacía tres años por vómitos, asociados a valores elevados de glucemia y acidosis metabólica, interpretada como diabetes mellitus tipo I, de inicio con cetoacidosis diabética. En aquella ocasión presentó un cuadro clínico de deterioro súbito del estado neurológico, se planteó un edema cerebral que se demostró por tomografía axial computarizada (TAC) de cráneo, situación que fue interpretada por el facultativo como consecuencia del tratamiento impuesto. Dada la mejoría clínica se trasladó a sala de Endocrinología, se logró retirar el tratamiento con insulina y se siguió en consulta.
Se recogió el antecedente de otros ingresos por vómitos en el transcurso de este tiempo entre el primer y último internamiento. Resultó relevante al examen físico que la niña se percibió como delgada y apática.
En esta ocasión que se reporta ingresó en el Servicio de Cirugía Pediátrica del Hospital Provincial Pediátrico Universitario José Luis Miranda, de Villa Clara por cuadro de vómitos y dolor abdominal. Se realizó laparoscopia que mostró signos inflamatorios a nivel de fosa ilíaca derecha y se interpretó como apendicitis aguda. Fue intervenida quirúrgicamente sin complicaciones inmediatas y fue egresada a las 48 horas.
El mismo día del egreso acudió nuevamente a cuerpo de guardia con cuadro febril agudo, deshidratación severa y toma sensorial marcada. Se ingresó en la Unidad de Cuidados Intensivos Pediátricos (UTIP) y dado los antecedentes se planteó inicialmente como infección relacionada a los cuidados sanitarios con sepsis severa que requirió ventilación mecánica. Como resultado del estado neurológico, se realizó tomografía que informó: pérdida de la relación sustancia gris-sustancia blanca dada por hipodensidad a nivel de los núcleos talámicos, globo pálido y a nivel de putamen bilateralmente; incluía, además, el área del mesencéfalo. El resto de las estructuras cerebrales se definían de características normales, el sistema ventricular normal y espacios subaracnoideos supra e infratentoriales normales. (Fig. 1).
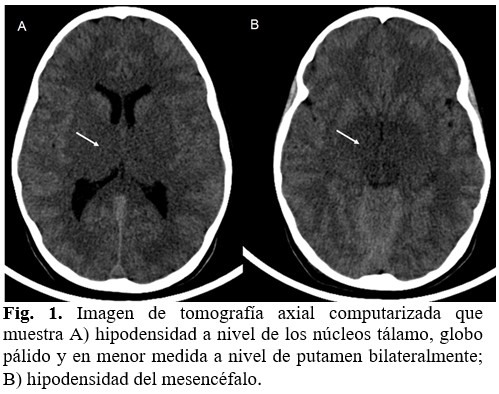
Se impuso tratamiento antiedema cerebral con mejoría del cuadro inicial, se trasladó de UTIP a los seis días de internamiento.
Se valoró por múltiples especialidades como Neurología, Endocrinología, Pediatría y Cirugía Pediátrica por considerar la sintomatología actual en relación con enfermedad metabólica y/o sepsis responsable del deterioro neurológico en relación con frecuencia al aporte de fluidos endovenosos.
Comenzó nuevamente con deterioro del estado neurológico y se consideró la posibilidad de encefalitis autoinmune por lo cual se impuso tratamiento con metilprednisolona y se trasladó a UTIP en estado de coma, con valoración de la escala de Glasgow de 8 puntos que llevó a la ventilación mecánica.
Se realizó discusión multidisciplinaria, se interrogó nuevamente a la madre que en esta ocasión aportó como dato relevante un fuerte olor en el sudor de la paciente al igual que las secreciones óticas y la orina. Se descartaron causas tumorales, reumatológicas, infecciosas y ante la sospecha de error congénito del metabolismo, trastorno del ciclo de la urea o aminoaciduria se indicaron nuevos exámenes complementarios, entre ellos la espectrometría de masa.
Se realizó nuevo estudio tomográfico de cráneo que mostró un empeoramiento de las condiciones neurológicas al reflejarse en la imagen hipodensidades difusas corticales y subcorticales, más acentuada en ganglios basales que demostraron la necrosis bilateral a ese nivel. Incluía, además, al tallo cerebral y núcleos dentados cerebelosos. (Fig. 2).
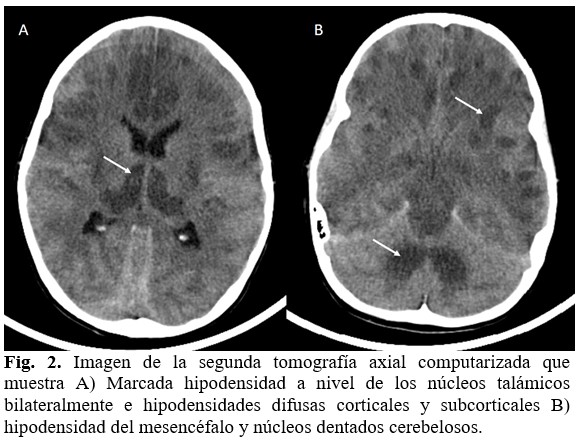
Existió nula mejoría a pesar del tratamiento, progresivamente profundizó el coma, se declaró en muerte encefálica y falleció.
Postmortem se recibieron resultados de exámenes complementarios, entre ellos el estudio en orina que demostró mediante cromatografía la elevación anormal de isoleucina, leucina y valina hecho que confirmó el diagnóstico de enfermedad de la orina con olor a jarabe de arce.
DISCUSIÓN
La EOJA es una enfermedad genética autosómica recesiva, cerebral degenerativa, causada por un déficit en la actividad de la deshidrogenasa de los cetoácidos de cadena ramificada, que provoca inadecuado almacenamiento de valina, isoleucina y leucina, tres aminoácidos esenciales de dicha cadena. Esta última y el ácido 2 oxoisocaproico son especialmente tóxicos para el sistema nervioso central.(2)
La acumulación de aminoácidos de cadena ramificada y de los alfa cetoácidos genera una neurotoxicidad severa que puede llevar a la muerte. La sospecha temprana de la enfermedad puede cambiar radicalmente la historia natural de la misma.(4)
Los estudios moleculares han permitido la localización en diferentes genes que codifican cada una de estas subunidades, lo que explicaría la heterogeneidad genética de esta afección, así como los desiguales fenotipos moleculares en función del locus afectado del complejo deshidrogenasa de los α-cetoácidos.(5)
La fisiopatología se debe principalmente a cuatro factores:(4)
- Alteración de la osmolaridad por acumulación de alfa cetoácidos (principalmente de Alfa isocaproato) y de leucina. Este último, un aminoácido osmóticamente activo. Con las fluctuaciones en la osmolaridad, la vasopresina incrementa la retención de agua. La consecuencia de esto será el edema cerebral.
- Neurotoxicidad y el disbalance de neutotrasmisores: a nivel de la barrera hematoencefálica (BHE) la leucina compite con el trasportador de aminoácidos LAT1, que explica muchos de los cambios neurológicos y psiquiátricos que se presentan a largo plazo en estos pacientes.
- Stress oxidativo e inestabilidad genómica: los hallazgos neuropatológicos incluyen desmielinización, incremento en la apoptosis y aumento del stress oxidativo.
- Daño en otras vías metabólicas: el alfa-ceto-isocaproato inhibe la piruvato deshidrogenasa, la alfa cetoglutarato deshidrogenasa y otros componentes de la cadena de electrones mitocondrial. Esto explica la hiperamonemia como hallazgo frecuente en estos pacientes.
Las manifestaciones clínicas están dadas por retraso psicomotor, problemas de alimentación y orina con olor a jarabe de arce.(2) Existen cinco fenotipos diferentes: clásica, intermedia, intermitente, sensible a tiamina y deficiencia de dihidrolipoil deshidrogenasa (E3). Estas se pueden diferenciar apoyándose en la edad de inicio, severidad de los síntomas clínicos, datos bioquímicos y respuesta al tratamiento con tiamina:(2,5)
- Forma clásica: es la forma más frecuente y es típica del recién nacido, aparecen los síntomas en el tercer o cuarto día después de la alimentación, con rechazo a la lactancia, letargia, alteraciones del tono muscular, convulsiones y coma.
- Forma intermedia: se diagnostica en etapa de lactante y prescolar (5 meses a 7 años), en esta forma los episodios de encefalopatía aguda son infrecuentes.
- Forma intermitente: suele presentarse con desarrollo prácticamente normal, en edad preescolar o en la adolescencia con eventos periódicos producidos por infecciones e ingesta alta de proteínas. Los síntomas más importantes son letargia, vómitos, ataxia, deshidratación, cetoacidosis e hipoglucemia. La elevación de los aminoácidos valina, isoleisina y leucina (VIL) en suero se detectan solo en los períodos de descompensación y en dependencia de la gravedad de la crisis pueden quedar secuelas neurológicas.
- Forma sensible a la tiamina: existe discordancia para su diagnóstico. Habitualmente estos pacientes no tienen la enfermedad aguda y su curso clínico es parecido a la forma intermedia, caracterizándose por un aumento de los aminoácidos, que se normaliza tras el tratamiento con dosis farmacológicas de tiamina.
- Deficiencia de dihidrolipoil deshidrogenasa (E3): es muy rara y las manifestaciones clínicas son similares a la forma intermedia.(5,6)
Dada la edad de la paciente, la ausencia de cuadros habituales, el desencadenamiento luego de la intervención quirúrgica, se sospecha que la intermitente fue la forma de presentación de este caso.
Ante las imágenes hipodensas en ganglios basales en espejo debe realizarse el diagnóstico diferencial con diversas afecciones como son:(1)
- Encefalomielopatía necotrizante subaguda (enfermedad de Leigh)
- Encefalopatías mitocondriales (MELAS, enfermedad de Kearns-Sayre)
- Aciduria glutárica tipo I
- Acidemia propiónica
- Acidemia metilmalónica
- Enfermedad de la orina con olor a jarabe de arce
- Deficiencia de sulfito oxidasa
- Enfermedad de Wilson
- Enfermedad de Hallervorden-Spatz
- Intoxicación aguda (monóxido de carbono, metanol, cianhídrico)
- Necrosis estriatal bilateral
- Hipoxia aguda severa (asfixia perinatal, ahogamiento, paro cardíaco)
- Traumatismo craneal
- Síndrome hemolítico-urémico
- Postencefalitis (parotiditis, echovirus)
Todos ellos van a estar representados por determinado cuadro clínico y/o resultados de laboratorio e imagenológicos (tomografía axial computarizada y resonancia magnética). En el caso que se presenta se evidenció la alteración de metabolitos en orina que avalaron el diagnóstico de EOJA y descartó otros tipos de acidemia u otras enfermedades incluidas en el diagnóstico diferencial.
La enfermedad de la orina con olor a jarabe de arce es una afección rara, con diversas formas clínicas. En el caso que se presenta se requirieron estudios complementarios de mayor complejidad, ante un cuadro clínico y evolución rara, que confirmaron este padecimiento. Los estudios de imágenes como tomografía computarizada ayudaron a evidenciar el daño neurológico, fue muy característica la hipodensidad de ganglios basales asociado a edema cerebral.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Contribuciones de los autores
Conceptualización: Yurisandra Jiménez González, Leidelén Esquivel Sosa, Yagima Fleites García, Yandy León de Armas.
Visualización: Yurisandra Jiménez González, Leidelén Esquivel Sosa, Yagima Fleites García, Yandy León de Armas.
Redacción- borrador original: Yurisandra Jiménez González, Leidelén Esquivel Sosa.
Redacción – revisión y edición: Yurisandra Jiménez González, Leidelén Esquivel Sosa, Yagima Fleites García, Yandy León de Armas.
Financiación
Hospital Provincial Pediátrico Universitario José Luis Miranda. Villa Clara. Cuba.
