INTRODUCCIÓN
La sirenomelia es una enfermedad congénita, extremadamente rara, llamada también simpodia, sympus, simelia, sirenomelus, monopodia, uromelia, feto cuspideo, síndrome de Mermaid, síndrome de Vater, anomalía de Duhamel o feto sirenoide. Ha llamado la atención desde la antigüedad. Se caracterizada por diversos grados de fusión, malrotación y disgenesia de las extremidades inferiores, similar a la cola de un pez, generalmente letales por la asociación de malformaciones complejas de carácter multisistémico.(1,2,3,4,5,6,7,8,9,10)
Tiene un incidencia de 1 en 100 000 nacidos vivos, ocurre con más frecuencia en embarazos monocigóticos que en embarazos simples y generalmente sólo uno de los mellizos está afectado, mientras que el otro es normal.(1, 2, 4, 5, 7, 8, 11)
Debido a la baja frecuencia con que esta entidad se presenta, para su mejor estudio y comprensión, se presenta este caso y una revisión de la literatura.
PRESENTACIÓN DEL CASO
Gestante de 42 años de edad, primigesta, sin antecedentes patológicos personales ni familiares de interés, ni exposición a factores teratógenos, que fue remitida con 17,5 semanas de amenorrea al servicio provincial de Genética Médica de Cienfuegos. La paciente refirió que antes de este tiempo no había asistido a los exámenes establecidos para el seguimiento de su embarazo.
En la exploración ecográfica se observó un feto único acorde a su edad gestacional, líquido amniótico disminuido en cantidad y placenta inserta en la cara posterior, de características normales, con estructuras óseas alteradas en ambos miembros inferiores describiéndose fusión del sacro a los iliums con fémur y tibia únicos y ausencia de ambos pies. Además, presentó ausencia de genitales internos y externos, vejiga, uréteres, uretra, recto, ano y arteria umbilical única. El diagnóstico planteado por el servicio de Genética Médica fue: sirenomelia acompañada de agenesia de genitales internos y externos, vejiga, uréteres, uretra, recto, ano y fusión del sacro con los íliums.
Se le brindó asesoramiento genético a la madre y esta decidió la interrupción de su embarazo realizándose una semana después. Se obtuvo cadáver en el que fue imposible determinar su sexo, superficie rojiza, al que se le realizó una necropsia clínica. A la exploración del hábito externo se observó desproporción entre la talla y la brazada, siendo esta última la mayor y corroborándose el resto de los diagnósticos planteados por el servicio de genética médica. (Fig. 1, Fig. 2 y, Fig.3).
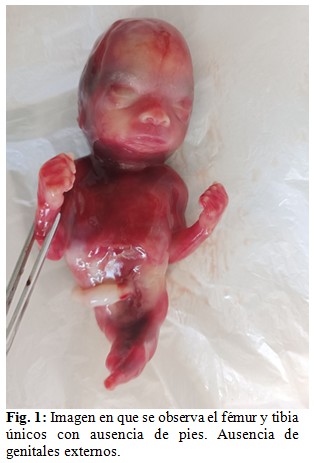
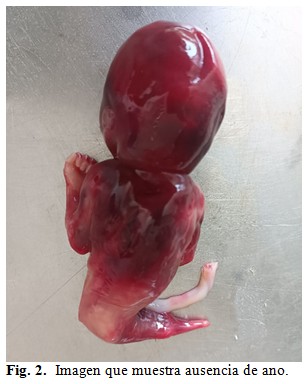
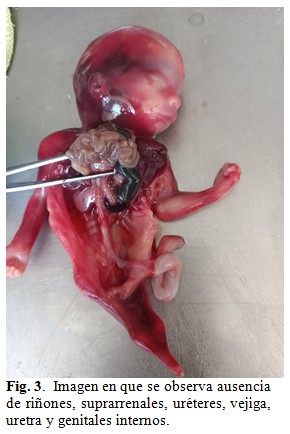
A la disección por aparatos se encontró porción rectal carente de luz y terminando en un extremo ciego. (Fig. 4).
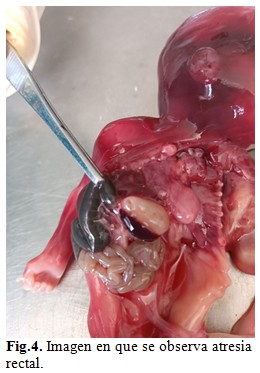
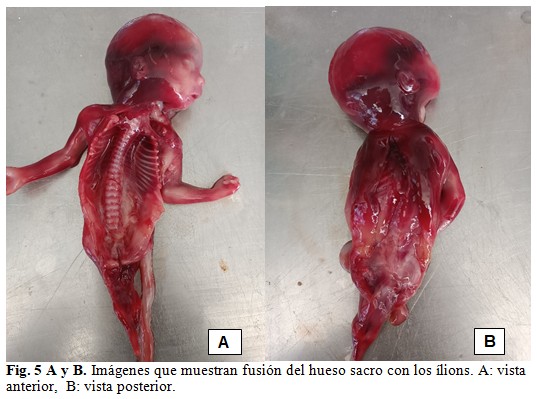
El estudio anátomo-patológico se concluyó como sirenomelia grado VI con fémur y tibia únicos y ausencia de pies, acompañada de fusión de sacro con huesos ilíacos, agenesia de genitales internos y externos, vejiga, uréteres, uretra, ano, arteria umbilical única y atresia rectal. Desproporción entre la talla y la brazada predominando esta última. (Fig. 5 A y B).
DISCUSIÓN
Esta afección se presenta a escala mundial y en todas las razas. Es más frecuente en los varones, con una frecuencia de masculino-femenino de 3:1 y en hijos de madre menores de 20 o mayores de 40 años,(2, 4,5,6,7, 8, 11) lo cual se cumple en este caso pues nuestra paciente tenía 42 años en el momento del diagnóstico, pero siendo portadora de un embarazo simple.
La etiología de esta anomalía es muy discutida y compleja, ya que en la mayoría de los casos se desconocen los antecedentes clínicos de la madre. Algunos autores refieren que esta malformación está asociada a diabetes materna, dosis excesiva de vitamina A, ácido retinoico, la exposición a la cocaína, ácido nalidíxico, anticonceptivos orales, insecticidas, humo de tabaco, radiación, metales pesados e hipovitaminosis(1, 2, 3, 5, 7, 8, 11) pero no se recoge ninguno de estos antecedentes en nuestra paciente.
Pero los factores de riesgo más relevantes asociados al síndrome de sirena, encontrados hasta el momento, son embarazos gemelares, en 10-15 % de los casos, principalmente, monocigóticos; la edad materna, principalmente, menores a 20 años o mayores a 40 años, y diabetes materna hasta en 35 % de los casos,(3, 7, 8) de los cuales nuestra paciente solo presentó una edad mayor de 40 años.
Todos los casos de sirenomelia informados han sido esporádicos y no se ha asociado a una alteración genética o hereditaria; aunque se sospecha una causa genética neomutacional dominante autosómica con predisposición masculina.(2, 3, 4, 7)
Existen múltiples opiniones acerca del mecanismo teratogénico y patogénico de esta malformación, pero aún en este campo no hay nada definitivo. Por esta razón han surgido muchas hipótesis, pero ninguna ha sido aceptada por unanimidad. Entre estas hipótesis se describen el fracaso en la rotación interna que normalmente ocurre en los miembros inferiores, la formación de adherencias o de pliegues estrechos en el amnios, la alteración temprana de las somitas caudales, que conlleva a anomalías del mesodermo y por lo tanto a alteraciones de los órganos que de él derivan; la teoría del déficit nutricional refiere que durante la tercera semana del desarrollo se produce agenesia del alantoides, con la subsecuente alteración de las arterias alantoideas que van a originar las arterias umbilicales definitivas.(1, 2, 3, 6, 7, 8)
Algunos autores denominan este conjunto de alteraciones de la porción caudal del cuerpo como síndrome de regresión caudal (CRS) y consideran que la sirenomelia es el grado máximo de este síndrome. Otra de las teorías que explica la aparición de esta anomalía es el desarrollo de un defecto temprano de la línea primitiva a nivel de los segmentos lumbares y sacros, lo cual puede ocasionar malformaciones de la cloaca, de los derivados del seno urogenital, así como también mal rotación de los miembros inferiores, los cuales rotan dorsalmente y pueden fusionarse. También puede alterarse la formación de la notocorda, ocasionando trastornos del desarrollo, tanto de la parte caudal del cuerpo, como del sistema nervioso. Otra de las teorías es la mecánica que plantea que el desarrollo caudal anómalo se produce por una fuerza intrauterina, que actúa en el extremo caudal del embrión.(1, 2, 5, 6, 9)
Se han propuesto varias clasificaciones para esta afección, la primera fue realizada en principio por Fórster (1861), dependiendo del número de pies: (a) apus, donde se encuentra fusión completa de las piernas y ausencia de pies; (b) unipus, presencia de un único pie; y (c) dipus presencia de dos pies. Posteriormente, Stocker y Heifetz (1987) lo clasificaron en siete tipos, dependiendo de la presencia o ausencia de los huesos de la extremidad inferior: (a) tipo I, con huesos del muslo y pierna formados adecuadamente; (b) tipo II, ausencia de formación del peroné de un lado; (c) tipo III, ausencia en la formación de ambos peronés; (d) tipo IV, fémures parcialmente fusionados con peronés fusionados; (e) tipo V, fémures parcialmente fusionados; (f) tipo VI, fémur y tibia únicos y (g) tipo VII, fémur único sin tibia.(1,2,3,4,5,6,7,8,9,12) En nuestro caso el feto puede ser clasificado como sirenomelia tipo apus según Forster y tipo VI según Stocker y Heiffetz.
La característica principal de los sirenomelos es la fusión de los miembros inferiores, que puede variar desde una fusión completa, hasta una unión membranosa de las piernas. Los peronés ocupan una posición interna y posterior a las tibias, las rodillas se doblan en sentido anterior y la planta del pie se dirige hacia delante. En algunos casos puede haber ausencia de la tibia. Puede verse como anomalía aislada pero con frecuencia se asocia a otras malformaciones como alteraciones de las vértebras lumbares, agenesia del sacro, fusión de los ilíacos, agenesia renal, de uréteres de vejiga y uretra. Las gónadas están presentes, pero el resto de las estructuras genitales raramente se observa o son ambiguos en un 80 % de los casos, cuando hay gónadas masculinas éstas se encuentran generalmente asociadas a un patrón cromatínico sexual femenino. La parte del cuerpo que se ubica por encima del cordón umbilical generalmente se desarrolla normal. La formación de la aorta y de las arterias umbilicales está alterada y casi siempre existe una sola arteria umbilical. Algunos sirenomelos pueden presentar reducción exagerada de la longitud de los miembros inferiores fusionados, con un aspecto de muñón. Estos casos se asocian con alteraciones del desarrollo del tronco.(1, 2) Se ha descrito la secuencia VACTERL (malformaciones vertebrales, atresia anal, cardiopatías, fístula traqueoesofágica, atresia esofágica, anormalidades renales y de las extremidades).(2, 4,5,6,7,8,9,10, 11)
Como hallazgos incidentales, pueden observarse anomalías del tubo neural, cardíacas y gastrointestinales. Se han referido casos con raquisquisis, espina bífida, anencefalia, cebocefalia, holoprosencefalia alobar, onfalocele, cloaca persistente, ectopia de suprarrenales, agenesia del cuerpo calloso y exónfalos.(1, 2, 4, 5, 7, 9)
Nuestro caso, además de la sirenomelia grado VI con fémur y tibia únicos y ausencia de pies, presentó fusión de sacro con huesos ilíacos y agenesia de genitales internos y externos, vejiga, uréteres, uretra, ano, arteria umbilical única y atresia rectal. Desproporción entre la talla y la brazada predominando esta última.
El diagnóstico prenatal de la sirenomelia es muy importante, ya que por tratarse de un producto no viable, se podría plantear la interrupción oportuna del embarazo. En todo caso, debe respetarse siempre el deseo de los padres en el momento de tomar cualquier decisión.(3, 4)
El método utilizado para detectar este defecto en vida intrauterina es el ultrasonido entre la semana 11 y 14 de la gestación. Durante el primer trimestre, el feto está rodeado de suficiente cantidad de líquido amniótico que proviene casi en su totalidad de los amnioblastos de la membrana amniótica; por esta razón se hace más fácil la visualización de las alteraciones presentes. Durante el final del segundo trimestre del embarazo y comienzos del tercero, el oligohidramnios o anhidramnios existente en la mayoría de los casos, puede entorpecer el diagnóstico de la sirenomelia, así como también la observación de las anomalías asociadas.(1,2,3, 4, 7, 8, 11)
El diagnóstico de las características esqueléticas de los sirenomelos es más fácil realizarlo cuando existe un solo fémur y una sola tibia.(1, 4)
Algunos autores refieren que utilizando la ultrasonografìa transvaginal a la octava semana posmenstrual, pueden ser observados los miembros fetales. A la novena semana posmenstrual ya se pueden identificar el húmero y el fémur, mientras que el radio, el cúbito, la tibia y el peroné, sólo pueden ser vistos a la décima semana. Sin embargo, se consigue poca bibliografía en la que se indique el diagnóstico de la displasia esquelética, observada por ecosonografía en el primer trimestre del embarazo.(1, 4)
La utilización del Doppler a color ha permitido una mayor precisión en el diagnóstico de la sirenomelia y de las anomalías asociadas. Una gran arteria que emerge de la aorta abdominal, la cual cursa centralmente hacia el cordón umbilical, corresponde a la persistencia de una arteria vitelina y puede ser considerado un hallazgo patognomónico para el diagnóstico prenatal de esta entidad.(1, 2, 4, 7, 8)
Se mejora la sensibilidad de la ecografía bidimensional con el uso de la radiografía simple de abdomen en el segundo y tercer trimestre de gestación, la amnioinfusión, la embrioscopía a las 12 semanas y la resonancia magnética. La radiografía en la autopsia, permite la categorización en base a la deformidad del esqueleto.(2, 3, 7)
La ultrasonografía transvaginal en tercera dimensión (3D), al igual que la ultrasonografía 3D en movimiento, también han sido utilizadas con éxito en el diagnóstico de la sirenomelia. Estas permiten corroborar las imágenes obtenidas con otros sonogramas y dan información adicional sobre los movimientos fetales y, en general, sobre la condición y desarrollo del feto.(1, 2, 3)
A nuestra paciente solo se le realizó ultrasonido a las 17.5 semanas puesto que no acudió a las consultas de ecografía previamente planificadas.
Sobre todo, se hace necesario realizar una historia clínica minuciosa y muy completa, con el fin de conocer la posible etiología de esta patología, y así evitar el riesgo que se repitan estas alteraciones en nuevos embarazos.(4) El principal diagnóstico diferencial es con el síndrome de regresión caudal, que suele presentarse con deformidades más leves y un volumen normal de líquido amniótico. Debido a la agenesia renal bilateral, como ya mencionamos, suelen tener una facies de Potter. La fusión de las extremidades inferiores, en la sirenomelia, establece el diagnóstico diferencial. Otros trastornos que deberían descartarse son el síndrome de Frase y la asociación VATER.(8)
El tratamiento del recién nacido es quirúrgico con el objeto de corregir las anomalías gastrointestinales, genitourinarias y osteomusculares en los casos donde no exista agenesia renal y el compromiso pulmonar no sea severo. Esta entidad es generalmente fatal, y sólo se han descrito cuatro casos vivos en aproximadamente 300 informados.(7, 8)
Excepto por casos muy raros, la sirenomelia es una condición letal en el período perinatal, que obstaculiza cualquier intención de tratamiento, ya que solo el 1,0 % de los casos sobrevive a la primera semana de vida, aunque se registran casos que han sobrevivido más allá del periodo neonatal, incluso más de 10 años. Las causas de muerte, supervivencia y calidad de vida dependen de las malformaciones congénitas asociadas.(2, 3, 7)
Algunos autores consideran que es posible hacer algo de prevención primaria de los casos de sirenomelia, a través del diagnóstico preconcepcional apropiado y del tratamiento de todos los tipos de diabetes. Ello evita la exposición temprana del embrión a altos niveles de glucemia, con sus posteriores consecuencias graves. Según esta teoría, podría evitarse hasta 20 % de todos los casos de sirenomelia.(3)
Conflicto de intereses
Las autoras plantean que no existe conflicto de intereses.
Contribuciones de los autores
Conceptualización: Lilian Rachel Vila Ferrán, María Antonia Ocaña Gil.
Visualización: María Antonia Ocaña Gil.
Redacción del borrador original: Lilian Rachel Vila Ferrán, María Antonia Ocaña Gil.
Redacción, revisión y edición: Lilian Rachel Vila Ferrán.
Financiación
Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. Cuba.
