INTRODUCCIÓN
Las distrofias miotónicas se distinguen por ser enfermedades genéticas de herencia autosómica dominante, que cursan con alteraciones en los músculos y también multisistémicas. Se describen dos tipos principales de la enfermedad: enfermedad de Steinert o distrofia miotónica tipo I y la distrofia miotónica tipo 2, también llamada síndrome de Ricker; ambas tienen cuadros fisiopatológico y clínico muy similares; pero también existen diferencias muy marcadas entre ellas.(1)
Existen formas pediátricas, ya sean neonatales (Steinert congénito), muy graves, con síndrome malformativo, gran hipotonía, insuficiencia respiratoria, muerte frecuente, o infantiles (Steinert infantil), que se manifiesta por lentitud y problemas de aprendizaje que conducen al fracaso escolar y variable.(2)
No existen datos de la prevalencia de esta enfermedad en Cuba y no se encontró en base bibliográfica para América Latina.
En términos clínicos la distrofia miotónica tipo 1 o enfermedad de Steinert puede clasificarse en cuatro diferentes subtipos: a) suaves, b) clásicos o de la edad adulta, c) juveniles y d) tipo congénito.(3)
Forma suave: también llamada por otros autores como de inicio tardío oligosintomática; se distingue por síntomas leves, como cataratas prematuras y calvicie, características clínicas únicas. Puede aparecer una miopatía de inicio tardío y la miotonía puede ser solo detectable por electromiografía. Pueden surgir alteraciones de la conducción cardiaca, dando lugar a una vida más corta.
Clásica o de la edad adulta: la edad de inicio suele ser en la segunda o tercera décadas de la vida. Los síntomas más frecuentes son la debilidad distal, que implica los flexores largos de los dedos de los brazos y las dorsiflexores de las piernas, lo que da lugar a los síntomas relacionados con la fuerza de la mano de agarre y mayor incidencia de tropiezo. Además, los pacientes pueden tener cataratas y calvicie y las alteraciones de la conducción cardiaca están presentes en forma regular. Existe miotonía clínica, también pueden ocurrir síntomas gastrointestinales y fatiga. La apatía, falta de iniciativa, la somnolencia diurna y la fatiga pueden ser síntomas importantes. Estas características tienen un efecto significativo en la calidad de la vida.
Forma congénita: la forma más grave de distrofia miotónica congénita tipo 1 se manifiesta de manera prenatal mediante la reducción de los movimientos fetales, polihidramnios y varias deformaciones detectadas en la ecografía fetal.
Algunos autores consideran tres síntomas principales o cardinales: debilidad muscular, miotonía y cataratas.
La miotonía se manifiesta habitualmente como rigidez, que los padres notan desde la edad escolar hasta la segunda o tercera década de la vida.
Por no existir datos sobre la prevalenia en Cuba y ser poco frecuente esta entidad nosológica, se decidió la presentación de los casos.
PRESENTACIÓN DE LOS CASOS
Caso 1
Se trata de un paciente de 15 años (Figura 1) con antecedentes de salud aparente hasta los 12 años cuando los padres comienzan a notar alteraciones del lenguaje, haciéndose este monótono y con un empeoramiento lentamente progresivo y sin afectaciones en las funciones corticales superiores, con un desarrollo escolar normal hasta ese momento.

El paciente se mantuvo con una evolución progresiva del cuadro con compromiso de la deglución, caída de parpados bilateral y de forma simétrica y comienza entonces con caídas frecuentes, por lo que acudió a consulta de neurología. Además el paciente comienza a quejarse de alteraciones visuales por lo que acude a oftalmología, donde se le diagnosticó la presencia de miopía.
Al examen físico se encontró la presencia de fenómeno miotónico en las extremidades superiores, debilidad en las cuatro extremidades, la presencia de calvicie prematura con debilidad y atrofia de los músculos temporales y maseteros, sin alteraciones cardiovasculares evidentes hasta el momento del examen.
Se interpretó como la forma infanto juvenil, en que los niños sufren debilidad muscular, pérdida de la fuerza o miotonía, además de dificultades en la escuela, que generalmente son por alteraciones visuales y del lenguaje, por lo cual generalmente suelen buscar a un neurólogo por posible retraso mental.
Al paciente se le realizó también electromiografía que mostró la presencia del fenómeno miotónico.
Caso 2
Durante la anamnesis en el caso anterior, se evidenció en la madre que esta presenta también signos y síntomas distintivos como: calvicie, atrofia de músculos faciales fundamentalmente temporales y la presencia del fenómeno miotónico en ambas manos, cuadro que inició cuando tenía aproximadamente 30 años de edad, por lo que se le indica la electromiografía dando resultados similares a los de su hijo.
No se recogen otros antecedentes familiares de la enfermedad en la familia. (Figura 2).
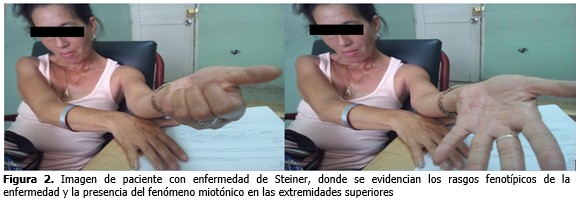
A la madre en este caso se le realizó EMG donde también se demostró miotonía, así como alteraciones de la conducción. Se diagnosticó con enfermedad de Steinert a dos miembros de la familia, en este caso madre e hijo. La madre comenzó con síntomas desde 1989, con problemas relacionados con debilidad muscular progresiva, habituales de los miembros inferiores o cintura pélvica y problemas de conducción cardiaca.
DISCUSIÓN
En general, las distrofias miotónicas son objeto de una amplia investigación debido a su importancia clínica e intrigante biología molecular. La degeneración progresiva de los músculos que lleva a la debilidad incapacitante y pérdida del tono muscular, en combinación con afectación multisistémica, son las principales características de la distrofia miotónica tipo 1 (también conocida como enfermedad de Steinert) y la distrofia miotónica tipo 2.(3-5)
En múltiples casos, la verdadera naturaleza de la enfermedad no se entiende hasta que a uno de los padres, generalmente la madre, se le diagnostica distrofia miotónica tipo 1 en la edad adulta como sucede en nuestro caso.
Epidemiología
Antes de la identificación de las distintas mutaciones genéticas, la prevalencia combinada de las distrofias miotónicas se estimó en 1 por cada 8,000 (12.5/100,000), sobre la base de comprobación clínica. Sin embargo, las estimaciones de prevalencia pueden variar ampliamente según la población.(6,7)
Características clínicas.
Existen dos tipos de distrofias miotónicas: la tipo 1 o enfermedad de Steinert, que puede subdividirse en cuatro subgrupos clínicos, y la tipo 2, conocida también como miopatía miotónica proximal o síndrome de Ricker. A pesar de las características de diagnóstico básicas comparables y de los múltiples órganos en común participación, hay características clínicas específicas de ambos tipos que les permiten ser distinguidos.(1,8)
En los pacientes con distrofia miotónica tipo 1 la debilidad muscular esquelética lleva a la inmovilidad y esta, a su vez, a insuficiencia respiratoria, disartria y disfagia, que es la causa principal de discapacidad grave y de muerte en las últimas etapas de la distrofia miotónica tipo 1.(9,10)
La debilidad muscular afecta la cara, el cuello y los músculos de las extremidades distales en paralelo con pérdida de masa muscular. Existe atrofia en el músculo temporal que, junto con la ptosis, contribuye al característico aspecto facial miopático, que se destaca por calvicie frontal en los hombres.
Invariablemente se encuentra miotonía en la presentación adulta de la distrofia miotónica tipo 1, en los datos clínicos y en la electromiografía. El signo más común es miotonía por percusión en el músculo tenar y, con menor frecuencia, la miotonía por agarre.
Las cataratas se detectan con exámenes oftalmológicos y el tratamiento de elección es la extirpación quirúrgica de las mismas.
Otra de las principales manifestaciones de la distrofia miotónica son los trastornos de la conducción, entre ellos los bloqueos auriculoventriculares, de rama y hemifasciculares.(3,11-13)

Formas clínicas
Forma juvenil: los pacientes no tienen los síntomas musculares característicos; los niños sufren debilidad muscular, pérdida de la fuerza o miotonía. En cambio, tienen dificultades en la escuela, que generalmente hacen buscar un neurólogo pediatra por retraso mental.
Al igual que con la enfermedad de tipo congénito 1, los niños con inicio de distrofia miotónica tipo 1 en la infancia padecerán síntomas musculares a una edad mayor, con discapacidades físicas comparables con las de la forma severa del adulto.
Forma congénita: la forma más grave de distrofia miotónica congénita tipo 1 se manifiesta de manera prenatal mediante la reducción de los movimientos fetales, polihidramnios y varias deformaciones detectadas en la ecografía fetal.
Al nacer, los bebés tienen graves problemas de hipotonía en las extremidades, el tronco, respiratoria y facial, lo que lleva a insuficiencia respiratoria y dificultades para la alimentación.
Con cuidados intensivos los niños sobreviven y no necesitan ventilación asistida, algunos de ellos pueden llegar a caminar y retrasar el daño muscular hasta la segunda década de la vida.
Los pacientes con distrofia miotónica tipo 2 tienen cuadros clínicos diversos. El fenotipo clínico es muy variable: discapacidades a la edad de 40 años en adelante, muerte cardiaca temprana, debilidad proximal leve que es apenas reconocible y concentraciones ligeramente elevadas de creatinincinasa en pacientes de edad avanzada.
El primer síntoma subjetivo es, generalmente, la debilidad de la extremidad inferior proximal, que causa dificultades para subir escaleras o dolor. Las características cardinales de la distrofia miotónica tipo 1, como la miotonía, pueden estar ausentes en pacientes con el tipo 2 de la enfermedad, incluso, los cambios en la electromiografía y las cataratas se observan en pocos individuos en el momento del diagnóstico.
La debilidad muscular en los pacientes con el tipo 2 comienza en una etapa posterior, el curso clínico es más favorable y la vida es casi normal.
Diagnóstico
Estudios sanguíneos. La actividad de la creatinincinasa en suero es, por lo general, ligera o moderadamente elevada en los pacientes con distrofia miotónica tipo 1 o 2, aunque las mediciones normales de esta enzima son frecuentes en la enfermedad tipo 2. El aumento de las concentraciones de las enzimas hepáticas, en particular γ-glutamiltransferasa, es hallazgo común, al igual que hipogammaglobulinemia por IgG por razones hasta ahora desconocidas.
La electromiografía es una de las pruebas de elección, muestra la combinación de la miotonía y cambios miopáticos considerados patognomónicos para el diagnóstico de distrofia miotónica.
Tratamiento
Hay diversos enfoques terapéuticos de la distrofia miotónica tipo 1 que no se basan en la modificación de la enfermedad, sino en orientar la gestión exclusivamente sintomática. La mexiletina y algunos antiepilépticos son una alternativa de tratamiento, modulan los canales de sodio y, por tanto, disminuye la miotonía y la fatiga.(11,14)
Conflicto de intereses:
No conflicto de intereses.
Contribución de autoría:
Dra. María Octavina Rodríguez Roque 40 %
Dr. Julio López Argüelles 10 %
Dra. Ada Sánchez Lozano 10 %
Dra. Didiesdle Herrera Alonso 10 %
Dra. Leydi M. Sosa Águila 10 %
Dr. Yansel Rodríguez Ramírez 10 %
Financiación:
Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos.
