INTRODUCCIÓN
La histiocitosis de células de Langerhans (HCL), también conocida como histiocitosis x, granuloma eosinofìlico, enfermedad de Letterer – Siwee y enfermedad de Hand – Schüller-Chistian. Es una enfermedad proliferativa que se caracteriza por la acumulación irregular o localizada de células en cualquier lugar del organismo.(1)
En 1913 se empleó por primera vez la palabra histiocito y desde entonces su sistema celular ha recibido diferentes nomenclaturas. En 1953 Lichtenstein observó que la histología de las lesiones eran muy parecidas y concluyó que representaban la misma enfermedad con distintas pautas de afección orgánica, por tanto, este autor englobó las tres entidades bajo la denominación de histiocitosis X.(2,3)
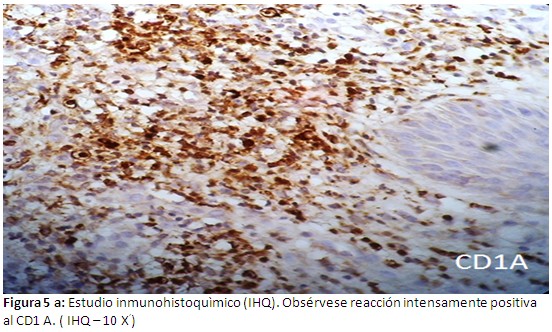
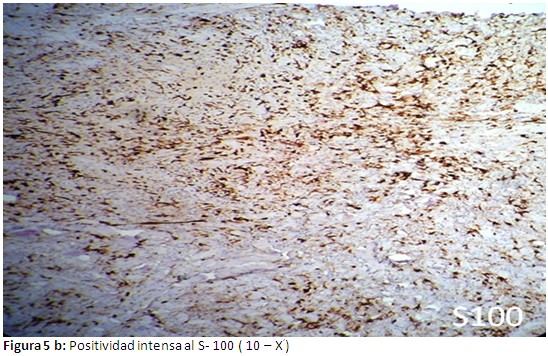
Se puede afirmar que la HCL (histocitis de células de Langerhans) es una enfermedad poco frecuente que se caracteriza por la acumulación y proliferación de histiocitos, eosinofilos y células de Langerhans, con inclusión de gránulos de Birbeck detectables por microscopia electrónica, afectando órganos y sistemas de forma aislada o múltiple. El diagnóstico se establece mediante la biopsia de la lesión y la confirmación por estudios inmunohistoquìmicos con reacción intensamente positiva al CD 1 A , CD 207 y S - 100. Las distintas formas de presentación originan un enfoque terapéutico y pronóstico diferente, que van desde las formas benignas, auto limitadas, con resolución espontánea hasta otras de curso tórpido o maligno.(4-6)
La HCL es una enfermedad de etiología desconocida, se cree que se trata de un proceso auto inmunitario en el cual las células involucradas atacarían el organismo, en lugar de combatir las infecciones. Es infrecuente en adultos, con una incidencia global estimada de 8 – 9 casos por millón de niños, su presentación clínica es muy heterogénea con un amplio rango, desde una lesión solitaria a formas graves multifocales y diseminadas. Los pacientes con compromiso de un solo órgano en general tienen un pronóstico favorable. La afectación ósea es frecuente, pero la afectación vertebral es rara.(7,8) Entonces al paciente se le diagnóstico una HCL a forma poliostótica, y se mostró importante compromiso vertebral y otras lesiones óseas localizadas a diferentes niveles.
PRESENTACIÓN DEL CASO
Se presenta un caso de paciente blanco masculino de 39 años de edad con antecedentes aparentes de buena salud. Acude a consulta con molestias y dificultad para caminar con pérdida de la fuerza muscular en ambos miembros inferiores, dolores óseos de moderada intensidad de carácter intermitente (posteriormente se hicieron constantes) que se localizaban en el tórax, región cervical y extendiéndose hasta el territorio lumbo sacro. No se aliviaba con analgésicos habituales y además notó un aumento de volumen en la región frontal derecha de aproximadamente 4 cm de diámetro, que no era doloroso al tacto. Por todo lo anterior decide solicitar asistencia médica, en principio llega remitido a nuestro hospital e ingresado en la sala de oncológica clínica, con el diagnóstico preliminar de mieloma múltiple y síndrome de compresión medular con el objetivo de recibir radioterapia y aliviar los síntomas. Es sometido a 10 sesiones de radioterapia, a razón de 3Gy por sesión, recibiendo una dosis tumoral total de 30 Gy mejorando parcialmente las manifestaciones clínicas. Se practicaron estudios tomográficos, luego se toman muestras de la lesión tumoral de la frente para continuar estudios. Se llegó a la conclusión que el paciente padecía de histiositosis de Langerhans a forma poliostótica. Actualmente continúa recibiendo quimioterapia, con discreta evolución clínica y pronóstico reservado.
Datos positivos del paciente:
Dolor en región lumbo sacra y tórax que se exacerba a la exploración de los movimientos articulares, paraparesia a nivel de miembros inferiores, lesión tumoral de unos 4 cm de diámetro en la región derecha del hueso frontal, de consistencia dura no dolorosa de bordes regulares, de color blanco amarillento. (Figura 1).
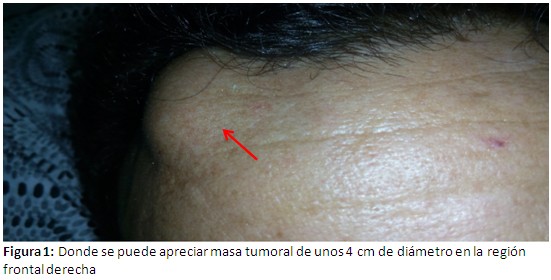
Exámenes complementarios:
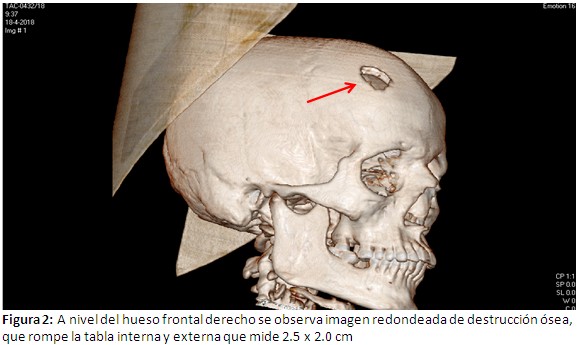
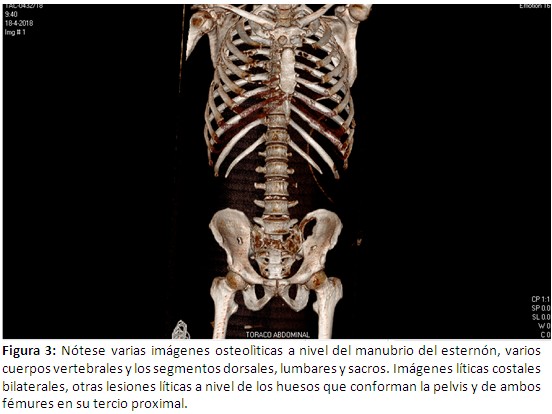
Hematocrito: 44% ; leucocitos 13.3 x109 U/L ; polimorfos: 76% ; linfocitos: 18% ; monocitos 5% ; eosinofilos 1% ; plaquetas: 255 x 109 U/l ; eritrocedimentaciòn globular 98 mm/h ( valor normal de 3 a 10 mm/h); resto de hemoquìmica, estudios enzimáticos de hígado y páncreas, función renal, iones, VDRL y VIH sin alteraciones; ultrasonografía abdominal y radiografía de tórax : normal; tomografía axial computarizada (TAC) simple de cráneo: a nivel de hueso frontal derecho se observa imagen de destrucción ósea que rompe la tabla interna y externa que mide 2.5 x 2.0 cm, (Figura 2) TAC toraco –abdominal – pelvis: imágenes osteolìticas a nivel del manubrio esternal, varios cuerpos vertebrales de los segmentos dorsales, lumbares y sacro, imágenes líticas costales bilaterales, también a nivel de los huesos que conforman la pelvis y de ambos fémur en su tercio proximal; resto del examen sin alteraciones. (Figura 3).
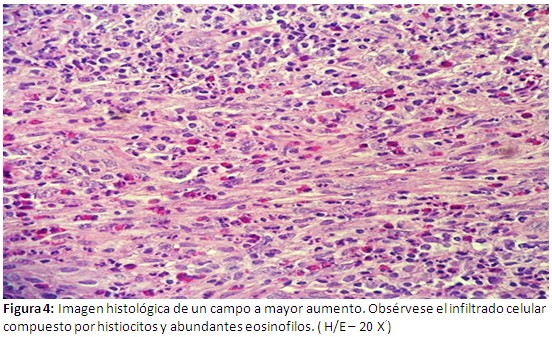
Informe de estudios histopatológicos, infiltrado celular compuesto por histiocitos y abundantes eosinofilos (Figura 4). En las evidencias inmunohistoquìmicas, se puede probar una reacción intensamente positiva al CD1-A (Figura 5-a) y positividad intensa al S-100 (Figura 5-b). Se concluye el caso como una Histiocitosis de células de Langerhans a forma poliostótica.
Conducta terapéutica: El tratamiento se indicó por decisión de un grupo interdisciplinario y de acuerdo a las normas de oncología, al recibir prednisona, vinblastina, 6 mercaptopurina y metotrexate, todas a las dosis recomendadas, la respuesta a la terapéutica hasta la fecha se puede considerar como limitada.
DISCUSIÓN
Las células de Langerhans fueron descritas por primera vez en el año 1847 por Poul Langerhans. Su principal función es presentar antígenos para linfocitos T y se encuentran en el estrato suprabasal de la epidermis y la dermis, membranas mucosas incluyendo la superficie ocular, linfonodos, timo, bazo y tejido óseo; se afecta con mayor frecuencia el cráneo, costillas, vértebras y mandíbula, esta última se puede encontrar comprometida entre un 73 – 75 % de los casos, y se presentan usualmente las lesiones en la zona posterior del cuerpo mandibular.(9) Varios autores plantean que la HCL es más frecuente en los niños entre el primer y tercer año de vida, aunque reconoce que puede aparecer a cualquier edad, con un leve predominio en los varones.(10,11)
Este paciente es un adulto joven de 39 años de edad, en el cual se evidencian lesiones óseas múltiples y son más evidentes las lesiones a nivel del cráneo específicamente en el hueso frontal, también se pueden apreciar las lesiones osteolíticas a nivel de múltiples costillas, vértebras , el sacro y ambos coxales. Aparece como un proceso denso y difuso de la unión dermoepidérmica, la muestra fue tomada de la lesión ubicada en la región frontal derecha .Otras imágenes histológicas comprueban un infiltrado celular compuesto por abundantes histiocitos y eosinofilos. Al realizar los estudios inmunohistoquìmicos correspondientes, se puede comprobar una intensa reacción positiva al CD1A, y se deja notar una positividad extrema al S-100. Estos hallazgos inmunohistoquìmicos coinciden con trabajos realizados por otros autores.(12,13) Se debe señalar que en nuestro paciente no se demostró lesión visceral alguna.
Existen variantes sindrómicas que pueden superponerse: forma localizada con afectación unifocal ósea o pulmonar: (granuloma eosinófilo [70%] ), más frecuente en niños y adultos jóvenes y de forma diseminada que da manifestaciones como: afectación multifocal ósea, con mayor afección craneal (región petrosa, base de cráneo, órbitas y maxilar), afectación extraesquelética del sistema reticulo-endotelial (hepatoesplenomegalia, linfadenopatías, manisfestaciones cutáneas, afectación del timo, tracto gastrointestinal, cerebral, pulmonar y renal. Se pudieran mencionar dos formas de presentación: crónica recurrente (Hand Schüler-Christian [20%] ). Este tipo es más frecuente en niños entre 1-5 años y aguda fulminante: Letterer-Siwe (10%), recurrente en niños menores de 2 años.
Se debe considerar factores de mal pronósticos como son: la extensión de la enfermedad y la edad de presentación.(12 -14) El paciente fue evaluado por una comisión multidisciplinaria y se encuentra recibiendo tratamiento según los normas oncológicas establecidas al efecto, con pronóstico reservado.
CONCLUSIONES
La HCL es una enfermedad rara, que abarca un amplio espectro de manifestaciones clínicas y daña a varios órganos o sistemas; por el contrario ser localizada, es más frecuente en los niños y adultos jóvenes. Su etiología aún no se define con claridad, sí esta demostrada una proliferación anómala de las células de Langerhans. Los hallazgos radiológicos iniciales son confusos y difíciles de interpretar, se requieren de avanzados estudios inmunohistoquìmicos para confirmar el diagnóstico. El pronóstico es variable y dependerá de la extensión de la enfermedad y la edad de presentación.
Conflicto de intereses: Los autores declaran no tener conflictos de intereses.
Contribución de autores: Los autores participaron en la redacción del trabajo y análisis de los documentos.
Financiación: Hospital Oncológico María Curie.
