INTRODUCCIÓN
Los significados de los términos ciencia y tecnología han variado significativamente de una generación a otra. Sin embargo, se encuentran más similitudes que diferencias entre ambos términos.
La ciencia (y mucho más la tecnociencia) no es solo una actividad teórica, es una actividad social, institucionalizada, portadora de valores, cultura. Es un proceso social profundamente relacionado con la tecnología, lo que acentúa la influencia sobre ella de muy variados intereses sociales, económicos, políticos, entre otros. Las fuertes interacciones entre ciencia, tecnología e intereses impiden disociar la ciencia de sus metas e impactos.1
La presencia progresiva de la experimentación a partir del siglo XVII y la complejidad creciente de los recursos y habilidades técnicas que ella reclama, determinan que la relación del investigador con los procesos que estudia esté cada vez más mediada por toda una extensa red de dispositivos tecnológicos. Lo que se puede investigar y las conclusiones que es posible alcanzar sobre los procesos estudiados con frecuencia es altamente dependiente de la tecnología disponible.1
Cada día se ha hecho más claro que la ciencia y la tecnología son procesos sociales profundamente marcados por la civilización donde han crecido; el desarrollo científico y tecnológico requiere de una estimación cuidadosa de sus fuerzas motrices e impactos y un conocimiento profundo de sus interrelaciones con la sociedad.1
En salud se puede observar una relación cada vez más estrecha entre ciencia y tecnología, la denominada tecnociencia1,2 donde desaparecen los límites plausibles entre ambas. Sus conceptos se han limitado a describir o explicar el fenómeno salud individual, lo que no ha permitido la salida de la visión orgánica, y ha dificultado por un lado la apreciación de los factores subjetivos y psicológicos que intervienen en él, y por otro, la mirada individualizada, la que no ayuda a clarificar lo social en salud.
No se trata solo de velar por los impactos sino de tomar nota de la dirección social que sigue la tecnociencia, en particular el problema de la apropiación privada del conocimiento, y discutir en qué medida ese desarrollo afirma o cuestiona el alcance humanitario de la ciencia, de explicar cómo la misma se expresa en su exploración, de la evolución histórica de su objeto, de sus condicionantes sociales de diversa índole (políticas, económicas, ideológicas, culturales), y también debe ser capaz de valorar sus resultados sociales, lo que significa la debida contextualización de su investigación.1
En la actualidad son incontables los ejemplos que ilustran la naturaleza de la tecnociencia, entre ellos tenemos la química sintética, la biotecnología y la farmacología,1 esta última encargada de demostrar científicamente, a través de técnicas y métodos, que una sustancia tiene actividad farmacológica y efectos indeseables aceptables para el hombre. Esta rama de las ciencias médicas se divide en la farmacología preclínica encargada de estudiar el efecto y la toxicidad en animales de experimentación (evaluación preclínica) y la farmacología clínica que estudia la eficacia, seguridad y características farmacocinéticas de los medicamentos en el hombre, utilizando como modelo experimental el ensayo clínico.
El ensayo clínico es la metodología idónea para evaluar una terapéutica o intervención en humanos, comúnmente conocido como el estándar de oro,3,4 y constituye también un paradigma para el registro sanitario de tecnologías sanitarias en general.
En la actualidad, uno de los mayores retos que enfrenta la Industria Médico Farmacéutica y Biotecnológica, una vez transcurrida la etapa de investigación preclínica, es justamente la etapa de evaluación clínica.5 Por lo cual este trabajo tiene como objetivo ofrecer una reflexión sobre los aspectos más significativos de los ensayos clínicos y su impacto en la sociedad.
DESARROLLO
I.-Investigación clínica
Con el advenimiento de la penicilina en los inicios de los años 40 y el descubrimiento de otros antibióticos en esa misma década, comienza una avalancha desenfrenada en la búsqueda e introducción de nuevos productos con el fin de incrementar el arsenal terapéutico. Se abría para la humanidad un camino en el que el avance de las ciencias iba a ser capaz de encontrar, gradualmente, un remedio para cada mal. La historia demostró que ese camino no estaba exento de riesgos y sucesivamente fueron apareciendo productos eficaces, pero tenían el inconveniente de producir efectos adversos, lo cual determinó la necesidad de establecer pruebas cada vez más complejas, sofisticadas, costosas y de larga duración para introducir un nuevo producto en el mercado.6
Esta serie de exigencias se puso de manifiesto, una vez más, con la llamada “tragedia de la talidomida” en Europa a comienzos de los años sesenta, durante la cual, se estima que como consecuencia del uso he dicho medicamento, deben haber ocurrido entre 10 000 y 20 000 nacimientos de bebés con graves deformaciones, las cuales afectaban sobre todo (pero no de manera exclusiva) el desarrollo de los miembros. Todo lo cual hizo que surgieran leyes regualtorias (Medicines Act, Inglaterra 1968; Buenas Prácticas Clínicas (BPC), Estados Unidos 1977; aunque ya anteriormente existía en este país la enmienda Kefauer-Harris, 1962). Estas leyes garantizaban la seguridad y la eficacia de los nuevos productos a introducir en el mercado.6
Todo esto hizo que se desarrollara la investigación clínica, definida como una investigación sistemática de la biología, la salud o la enfermedad humana que, realizada sobre las personas, implica un conjunto de actividades orientadas a probar una hipótesis, obtener unas conclusiones y de esta manera contribuir a la obtención de un conocimiento generalizable y útil para otros.7
El procedimiento aceptado para la realización de la investigación clínica es el ensayo clínico que se define, de acuerdo a la Conferencia Internacional de Armonización (ICH, por sus siglas en inglés International Conference Harmonisation) como “cualquier investigación en sujetos humanos dirigida a descubrir o verificar:6
- Los efectos clínicos, farmacológicos y/u otros efectos farmacodinámicos de un producto en investigación
- Identificar cualquier reacción adversa al producto en investigación (perfil de seguridad)
- Estudiar la absorción, distribución, metabolismo y excreción de un producto en investigación
- Establecer la eficacia del producto en investigación para una indicación terapéutica, profiláctica o diagnóstica determinada.
Muchos coinciden en señalar que el primer ensayo clínico planeado de la historia fuera tal vez el que realizó James Lind, cirujano de la marina británica, en el año 1747, con el objetivo de averiguar el mejor tratamiento contra el escorbuto.6
El término ensayo clínico aparece por primera vez en la revista Lancet en 1931 (publicación anónima) y coincide curiosamente con la constitución del Comité de Ensayos Clínicos por el Medical Researh Council del Reino Unido. Años más tarde, Sir Austin Bradford Hill, considerado en la actualidad el padre del ensayo clínico moderno, publica el primer libro sobre el tema, titulado “Principles of Medical Statistics”. El primer ensayo clínico controlado se realizó en 1946, dos años después del descubrimiento de la estreptomicina por el Medical Research Council británico para probar la eficacia de este medicamento en el tratamiento de la tuberculosis.6
En el año 1950 aparece por primera vez el término doble ciego, debido a un ensayo clínico realizado por el grupo de Harry Gold en pacientes con angina de pecho, aunque probablemente durante los años treinta este mismo grupo realizó un estudio debidamente enmascarado. También se plantea que los primeros en utilizar la técnica de simple ciego fueron Ferguson y col. en 1927 al desarrollar un estudio de una vacuna contra el catarro común comparada con suero salino.6
El concepto de aleatorización había sido introducido por Fisher en el año 1923, cuando lo aplicó en una investigación agraria. Amberson y col. publicaron en 1931 un ensayo clínico sobre la eficacia de la sanocricina en la tuberculosis pulmonar, empleando por primera vez un método de asignación aleatoria bien rudimentario.6
II.-Ensayos clínicos. Sus características
El ensayo clínico controlado aleatorizado enmascarado (ECA) es el diseño ideal frente al cual comparar todos los demás diseños: 8
- Un ensayo es clínico cuando cualquier tipo de experimentación planeada involucra pacientes con una condición médica dada con el objetivo de elucidar el tratamiento más apropiado de futuros pacientes similares o también métodos de prevención o diagnóstico.
- Es controlado porque involucra la comparación de efectos de tratamientos entre un grupo intervenido y un grupo que actúa como control, para intentar evitar el potencial de proveer una visión distorsionada de la eficacia y/o efectividad del tratamiento;
- el hecho de ser aleatorizado significa que los investigadores asignan la exposición sobre la base del azar, es decir cada sujeto que entra al estudio tiene la misma probabilidad de pertenecer a un grupo o a otro, produciendo además, que los grupos en estudio sean comparables con respecto a factores de riesgo conocidos y desconocidos;
- Finalmente, el que un ensayo sea enmascarado o “ciego” quiere decir que los pacientes, los médicos, los evaluadores u otros participantes en la investigación, no conocen la intervención a la que está sometido cada paciente, disminuyendo de esta forma la introducción de sesgos ya que la comparación de tratamientos puede ser distorsionada si el paciente y aquellos responsables del tratamiento y evaluación conocen cual tratamiento está siendo usado.8
El desarrollo de un nuevo medicamento es un proceso largo, el tiempo medio que se tarda en comercializar una nueva molécula oscila alrededor de los 10 años. En este proceso intervienen varias fases de investigación, las cuales se dividen en: fase preclínica y fase clínica,9 esta última comprende cuatro fases diferentes (fases I-IV).
- Fase I. Seguridad y tolerancia. Farmacocinética y Farmacodinámica
Constituyen el primer paso en la investigación de un producto en estudio en el hombre. Corresponden fundamentalmente a estudios de farmacocinética y farmacodinamia. Abarcan las primeras pruebas en humanos, normalmente en voluntarios sanos, para evaluación preliminar de tolerancia, evidencia de acciones farmacológicas, rangos y regímenes seguros de dosificación, absorción, distribución, metabolismo y excreción.
Proporcionan información preliminar sobre el efecto y la seguridad del producto en sujetos sanos o en algunos casos en pacientes (en el caso de cáncer y Sida), y orientan la pauta de administración más apropiada para ensayos posteriores.9
- Fase II. Efectos, seguridad, tolerancia y otros aspectos farmacológicos
Esta fase comprende la investigación clínica inicial del efecto del tratamiento. Se realiza con un número limitado de pacientes que padecen la enfermedad o entidad clínica de interés (alrededor de 200) para estudiar una actividad biológica específica, el control o la profilaxis de una enfermedad.9
- La fase II-a “temprana” refleja los estudios iniciales (estudios piloto) para recabar la primera evidencia de la eficacia.
- En la fase II-b “tardía” se diseñan los estudios para dar respuestas definitivas a preguntas sobre la seguridad del fármaco y su utilidad terapéutica, exigiendo una monitorización rigurosa de cada paciente.
Los estudios fase II pueden servir como un proceso de selección para elegir aquellos fármacos con verdadero potencial para ser desarrollados en fase III, proporcionar información preliminar sobre la eficacia del fármaco y suplementar los datos de seguridad obtenidos en la fase I. Estos estudios sirven también para determinar el rango de dosificación apropiado. Por lo general son ensayos clínicos controlados y con asignación aleatoria de los tratamientos. Los criterios de inclusión y exclusión son estrictos.
Es de señalar el concepto de validez interna, la cual no es más que la fiabilidad de los hallazgos del ensayo clínico en relación con la población misma de estudio. Por tanto, un ensayo correctamente realizado y con una población de estudio muy homogénea, debido a los criterios de selección estrictos, posee una buena validez interna, aunque sus resultados pueden no ser necesariamente extrapolados a la población diana. 9
- Fase III. Eficacia, seguridad y tolerancia
Incluye un amplio rango de ensayos en los que participan numerosos investigadores para valorar la eficacia y seguridad de un producto nuevo bajo condiciones similares a aquellas que se puedan esperar de dicho fármaco cuando se encuentre en el mercado y considerando las alternativas terapéuticas disponibles en la medicación estudiada. Se realizan en una muestra de pacientes más amplia (desde varios cientos hasta miles) y representativa de la población general a la que se destina el medicamento.
La seguridad sigue siendo uno de los objetivos principales. Los efectos tóxicos predecibles del fármaco en estudio tendrán que haber sido descubiertos en las fases I o II. La fase III debe establecer la incidencia de los efectos secundarios comunes e, idealmente, indicar qué tipo de pacientes tienen un riesgo especial para desarrollar efectos secundarios menos frecuentes.
En los estudios fase III el fármaco en investigación se compara con los tratamientos estándares preestablecidos, si los hubiera, bien sea placebo o terapias reconocidas, a fin de confirmar evidencia de eficacia relativa.
Es importante que las condiciones en que se realiza el ensayo se aproximen lo más posible a la situación real en que luego va a ser utilizado el fármaco evaluado. Lo anterior se conoce como validez externa. Se refiere a que los resultados del ensayo sean representativos y relevantes respecto a la población diana y a la facilidad de extrapolar o generalizar los mismos a pacientes de diferentes niveles del sistema sanitario. Estos estudios constituyen el soporte para la autorización del registro y comercialización de un fármaco a una dosis y para una indicación determinada.
Por ello deben de ser controlados (a ser posible doble ciegos) y aleatorizados, incluyendo un número suficiente de pacientes que permita demostrar la eficacia y seguridad comparativa entre los tratamientos en estudio. Una vez que se registra el producto en investigación se desarrolla, por lo general, una cuarta fase.
- Fase IV. Estudios posteriores al registro y fármacovigilancia
Corresponden a los estudios postmarketing, es decir, aquellos que se realizan con fármacos comercializados. Se llevan a cabo para efectuar la fármacovigilancia del producto incluyendo la detección de efectos secundarios a largo plazo, así como posibles efectos del fármaco sobre la patología en sí misma o estudios de morbilidad y mortalidad.
También se utiliza la fase IV para estudiar nuevas indicaciones del producto, nuevas formulaciones y formas de dosificación o la comparación con otros fármacos ya conocidos.9
La investigación clínica es esencial para mejorar la calidad de vida de la población y el bienestar general de la sociedad. Los avances en el conocimiento fundamental solo se traducirán en calidad de vida y en bienestar para las personas cuando puedan ser aplicados para mejorar la prevención, el diagnóstico y el tratamiento de las enfermedades.
III.-Conferencia Internacional de Armonización y buenas prácticas clínicas
En el marco de la Conferencia Internacional de Armonización surgieron las Guías de Buenas Prácticas Clínicas, que definen una serie de pautas a través de las cuales los estudios clínicos puedan ser diseñados, implementados, finalizados, auditados, analizados e informados para asegurar su confiabilidad.10
El proceso completo de los ensayos clínicos se realiza apegado a estas guías de buenas prácticas clínicas, las cuales contienen una mezcla de políticas, principios y procedimientos con calidad ética y científica internacional, para diseñar, dirigir, registrar e informar acera de estudios clínicos. Su cumplimento en los estudios de investigación clínica aseguran que los derechos, seguridad, métodos de colección de datos, registro de información, la documentación y el análisis estadístico están bien soportados, pero sobre todo sean creíbles. Por esta razón las agencias regulatorias las toman como guías para normar y regular los estudios clínicos. Con este código ético, científico y regulatorio se anticipa la protección del ser humano.11
IV.-La investigación clínica en Cuba
Las primeras investigaciones médicas en Cuba se realizaron en La Habana por el Dr. Tomás Romay Chacón, en el año 1804. Sus trabajos fueron réplicas de estudios anteriores en otros países, con el objetivo de probar la efectividad de la vacuna contra la viruela.12
A partir de 1879, el Dr. Carlos J. Finlay realiza investigaciones, con el objetivo de demostrar su entonces hipótesis de que la picadura de la hembra del mosquito, clasificado actualmente como Aedes aegypti, era la responsable de la transmisión de la fiebre amarilla.12 Estas investigaciones médicas en Cuba, que se realizaron durante siglo y medio, costaron algunas vidas de investigadores y voluntarios; no obstante, en ningún momento se aprobó una legislación que regulara esta práctica, ni tampoco que la prohibiera. Esto ocurrió también en el resto de los países del mundo.12
Antes de 1989 cuando se pretendía realizar un estudio que implicaba una intervención experimental diagnóstica o terapéutica, y se empleaba un producto nuevo, se sometía a la consideración del Ministerio de Salud Pública (MINSAP), para obtener que fuera aprobada su aplicación por primera vez en el humano.13
El desarrollo acelerado de la industria médico-farmacéutica y biotecnológica, así como el de las agencias regulatorias de medicamentos y equipos en el nuevo mundo, aumentó las exigencias para el registro de fármacos y productos biotecnológicos, lo cual obligó a Cuba a trazar una estrategia que contribuyera al registro y comercialización; y lograr lo antes posible, a partir del momento de la obtención del producto, su rápida introducción en la práctica médica.14
Uno de los objetivos de esta estrategia fue la organización y desarrollo de la actividad reguladora en el país, proceso cada vez más exigente y riguroso, con una aproximación sucesiva a los estándares científicos internacionales. Para ello se creó en 1989, mediante una Resolución Ministerial, el Centro para el Control Estatal de la Calidad de los Medicamentos,14 autoridad competente para la autorización del registro de medicamentos de uso humano, nacionales y extranjeros para toda Cuba, que ya en estos momentos tiene también entre sus funciones el registro de equipos y dispositivos médicos.
Simultáneamente a la reestructuración del marco regulatorio, se creó la infraestructura de evaluación de productos médico-farmacéuticos y biotecnológicos priorizados para el desarrollo: Química farmacéutica, Preclínica toxicológica y farmacológica.14
Dentro de esta infraestructura surgió la necesidad de una institución que permitiera la evaluación clínica de nuevos productos que por su carácter priorizado, requerían su rápido registro sanitario en el país y fuera de él. A fines de 1991, se creó una organización para el diseño y conducción de los ensayos clínicos, concebida como centro nacional y red de coordinación: el Centro Nacional Coordinador de Ensayos Clínicos (CENCEC).14,15
Su propósito es diseñar y organizar la ejecución de los ensayos clínicos que evalúan los diferentes productos para el registro y la comercialización de medicamentos, reactivos, productos biológicos, instrumental, equipos y procedimientos terapéuticos; así como la realización de ensayos comparativos epidemiológicos, todo ello en rigurosa observancia de las buenas prácticas clínicas y a un costo razonable.14
Un propósito importante, a partir de su creación, fue conformar una Red Nacional Coordinadora de Ensayos Clínicos, la cual garantiza la realización de ensayos clínicos multicéntricos en todo el país, a través de la aplicación de un único protocolo en varios sitios clínicos con el fin de aumentar la eficiencia, alcanzar los resultados a mayor velocidad y con los estándares de calidad requeridos. Todo lo cual fue posible por disponer de un sistema de salud único y contar con la infraestructura y preparación científica adecuada del personal de la salud en todas las provincias.14
El Centro Nacional posee coordinadores en cada provincia y 3 subcentros ubicados en las provincias de Villa Clara, Camagüey y Santiago de Cuba. Esta organización permite desarrollar el trabajo nacionalmente, con la participación de investigadores de varios hospitales del país.
El CENCEC y su red constituyen una organización integral, diferenciados del resto de las instituciones similares en el mundo (centros de investigaciones por contrato). Algunas características diferenciadoras son el alcance nacional, el compartir funciones de servicios científicos con investigación académica y de salud, así como el perfeccionamiento de los recursos humanos14
A la par de la creación de estos centros se establecieron las normas de Buena Práctica Clínica (BPC) y se crearon los primeros Comités de Revisión y Ética. Estas normas se reelaboraron en 1995 y en 1999 se armonizaron con las de la Conferencia Internacional sobre Armonización (ICH), fueron publicadas en el 2000, incluyendo un acápite especial sobre los Comité de ética.5 El cumplimiento de las BPC asegura por un lado la solidez científica del estudio y por el otro garantizar la solidez ética, además establecen pautas que garanticen que los datos que surjan de las investigaciones sean adecuadamente conservados y puedan ser verificados, independientemente del lugar en donde se realice el estudio.5,15
El desarrollo de la Biotecnología y la obtención de productos médico farmacéuticos desde la década de los 80, en Cuba, es bien conocido. Durante las décadas de los 80s y los 90s, surgieron en Cuba decenas de centro de investigaciones. La industria biotecnológica cubana se financia hoy con sus propias exportaciones. Estos productos han requerido ensayos clínicos para ser registrados y comercializados tanto dentro como fuera del país, los cuales se han realizados en las instituciones de salud del país.
La provincia de Cienfuegos forma parte de la Red Nacional de Ensayos Clínicos desde sus inicios en la década de los 90, el principal sitio clínico es el Hospital Universitario Dr. Gustavo Aldereguía Lima. Han estado involucrados en los ensayos clínicos múltiples servicios, dentro de los que se destacan Oncohematología, Urología, Nefrología, Coloproctología, Psiquiatría, Angiología, Dermatología, Neurología, Neonatología, Gastroenterología, Ginecología, Farmacia, Laboratorio clínico, Anatomía Patológica e Imagenología.
En 2014 los ensayos clínicos, en diferentes etapas de realización, alcanzaron los 51 con 1060 pacientes incluidos, los cuales se beneficiaron con los nuevos productos biotecnológicos. Son múltiples los productos y también los centros promotores, nacionales e internacionales, participantes, conveniados a través del CENCEC o en forma monocéntrica entre la institución productora y el sitio clínico.
Entre los ensayos con un mayor impacto en el territorio se encuentran los relacionados con el tratamiento de las enfermedades oncológicas. Se han ejecutado varios protocolos con productos líderes de centros promotores nacionales, como son el Centro de Inmunología Molecular (CIM) y el Centro de Ingeniería Genética y Biotecnología (CIGB) principalmente. (Tabla 1).
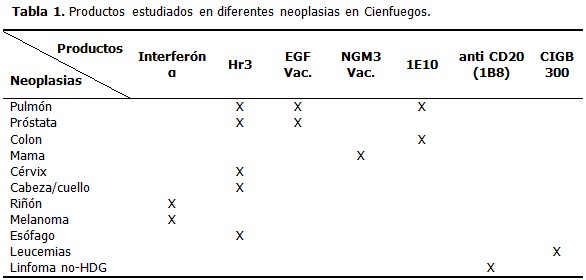
Otros ensayos se han realizado en el hospital con productos como:
- Surfacén en distrés respiratorio en el servicio de neonatología
- Supositorios de estreptoquinasa en trombosis hemorroidales
- Epocim en anemia en pacientes con insuficiencia renal y anemia posterior a la quimioterapia
- Leukocim en pacientes con neutropenias por quimioterapia
- Heberpro-P en pacientes con pie diabético
Además el hospital participó en el Proyecto de Ensayos Clínicos en Psiquiatría con la Universidad de Dalhouse, Canadá, cuyo objetivo fue elevar la capacidad de los investigadores en el diseño y conducción de ensayos clínicos, para evaluar terapias psicofarmacológicas, lo que además ha permitido elevar la calidad de la atención médica en esa área terapéutica.
Entre los productos más importantes se encuentran el Hr3 o Nimotuzumab, utilizado en la mayoría de las localizaciones anatómicas con proliferación de células neoplásicas, este producto está registrado en Cuba para tumores avanzados de cabeza y cuello, tumores astrociticos de alto grado de malignidad y tumores avanzados de esófago, así como el preparado vacunal EGF de amplio empleo en el cáncer de pulmón de células no pequeñas.
Se ha logrado llevar los ensayos clínicos hasta la atención primaria de salud (APS), con el uso en este nivel de atención de la vacuna terapéutica Cimavax-EGF contra el cáncer de pulmón avanzado de células no pequeñas, donde participaron policlínicos de 4 áreas del municipio de Cienfuegos y los policlínicos de Palmira y Cumanayagua. Se incluyeron 32 pacientes. En este nivel de atención se ejecuta, en conjunto con el Hospital Gustavo Aldereguía Lima, otro ensayo clínico en el cual los pacientes con cáncer de pulmón de células no pequeñas son tratados con Racotumomab o Nimotuzumab en comparación con el Docetaxel después de la primera línea de tratamiento oncoespecífico.
La ejecución de ensayos clínicos en la APS ha sido posible en Cuba debido al sistema fortalecido y bien estructurado de la asistencia médica, la Atención Primaria de Salud, que existe en todo el país, con la participación de un especialista de formación en Medicina General Integral y un equipo médico que trabaja en la promoción, prevención, curación y rehabilitación no solo del paciente, sino de la familia y la comunidad, abarcando de forma integral a todo los integrantes en su contexto sociocultural, al ser humano como ser social, y donde además se atienden a grandes grupos de pacientes, existiendo los registros donde se hallan bien caracterizados, reflejado en las fichas familiares e historias clínicas de los Consultorios Médicos de la Familia, lo cual ha sido una oportunidad y una rápida solución, ya que constituyen fuentes de posibles sujetos a incluir en las investigaciones, fundamentalmente en el logro del ritmo adecuado de inclusión necesario de los estudios.5
En la provincia se aplicó la vacuna contra el cólera en un ensayo clínico fase III, que demostró la seguridad y eficacia del producto, y la vacuna cubana contra el neumococo en más de mil niños desde uno hasta cinco años de edad, en ensayo clínico fase II/III.
Con el fin de poner a disposición de la población los mejores métodos profilácticos, terapéuticos y diagnósticos, una de las líneas estratégicas en nuestro país es la certificación de estos sitios para la realización de ensayos clínicos, lo cual garantiza la seguridad y eficacia de los medicamentos que se comercializan.16, 7 Debido a la experiencia del personal en el trabajo de ensayos clínicos así como la calidad de los ensayos realizados, el hospital se encuentra inmerso en el proceso de certificación de sitios clínicos, condición que, una vez alcanzada, le proporciona un mayor prestigio a la institución.17
Los ensayos clínicos mejoran los indicadores de salud de la población, que se modifican con la introducción de nuevos productos y producen cambios en los patrones de atención médica de la enfermedad en que se evalúa el producto, ya que se exigen para la aceptación del protocolo del ensayo clínico, los mejores estándares de diagnóstico, evaluación y tratamiento de la enfermedad; y la introducción de nuevos métodos para el diagnóstico, nuevas tecnologías de evaluación y nuevas terapéuticas asociadas al producto en estudio como los tratamientos concomitantes o tratamientos activos utilizados en los grupos controles.
CONCLUSIONES
La investigación clínica genera conocimiento que sirve para mejorar la salud y el bienestar y/o aumentar la comprensión de la biología humana. Los ensayos clínicos, como un paradigma de la investigación clínica, proporcionan innumerables beneficios para una asistencia médica de calidad. Producen cambios en indicadores de salud y garantizan mejores estándares de diagnóstico, evaluación y tratamiento de enfermedades.
Muchos productos ofrecen una alternativa de vida y salud en diversos campos, con una repercusión social importante, mejoran la calidad de vida y una mayor supervivencia en pacientes con enfermedades como el cáncer.
La ciencia es, en sí misma, una actividad social; en Cuba es evidente su impacto en aras de elevar la calidad de vida de sus habitantes y los de otros países, gracias al desarrollo de nuevos productos.
