INTRODUCCIÓN
El sarcoma de Ewing pertenece a las neoplasias de células pequeñas, azules y redondas.1 En la actualidad se considera que el sarcoma de Ewing y el tumor neuroectodérmico primitivo (PNET), descrito inicialmente como una entidad distinta, son parte de los denominados tumores de la familia del sarcoma de Ewing.2 Los sitios primarios donde principalmente se desarrolla son la pelvis ósea, fémur y huesos de la pared torácica.3
Se reporta una incidencia anual de 2,73 casos por cada millón de habitantes. Afecta a niños, adolescentes y adultos jóvenes. El 90 % ocurre en las dos primeras décadas de la vida. La media de edad se encuentra entre los 13 y 19 años, con un pico de incidencia entre los 5 y los 13 años. En Europa se encuentra en cinco de cada cien mil habitantes por año.4 Es la cuarta neoplasia ósea primaria, pero la segunda (después del osteosarcoma) en menores de 25 años y la primera en menores de 10 años. El 75 % de los sarcomas de Ewing se hallan localizados al momento del diagnóstico y entre el 25 y el 30 % debuta con metástasis, más frecuentes en pulmón y hueso.4,5
Se considera de interés la presentación de este caso por la localización poco usual del sarcoma en Ewing.
PRESENTACIÓN DEL CASO
Se presenta una paciente femenina, mestiza, de 37 años de edad, con antecedentes de asma bronquial controlada y ex fumadora. A causa de un tumor en la mano izquierda, de 11 meses de evolución, acompañado de dolor e impotencia funcional, se le había realizado biopsia incisional en otro centro hospitalario, en julio del 2014; este informó de un sarcoma sinovial. En septiembre de 2014, la paciente acudió al Instituto de Oncología y Radiobiología (INOR); mantenía los mismos síntomas descritos anteriormente. Se decidió su inscripción para ser tratada en el INOR. Fueron indicados estudios de extensión y exámenes prequirúrgicos. Se planteó como estadio cT1 N0 M0 Gx, etapa IA.
Examen físico
Se palpó en la mano izquierda lesión pétrea de más o menos 4 cm de diámetro, en la región tenar, sin signos inflamatorios, con cicatriz en la región dorsal, resultado de la intervención quirúrgica anterior.
Estudios imagenológicos
Tomografía axial computarizada (TAC) realizada el 14/10/2014, que informó lo siguiente: mano izquierda con lesión osteolítica en primer metacarpio izquierdo, con masa heterogénea adyacente de 39 x 35 mm y densidades que oscilan entre 41 y 50 UH. Sin alteraciones en la región torácica. (Figura 1).
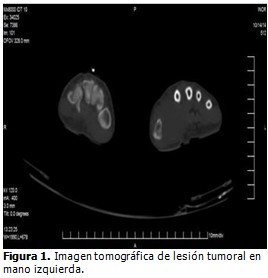
El 22 de enero de 2015, la paciente fue sometida a intervención quirúrgica, en la que le fueron amputados el primer y segundo dedos de la mano y primer metacarpiano.
El análisis anatomopatológico de la pieza quirúrgica informó sarcoma de células redondas de la familia del tumor Ewing/PNET, persistente en la primera falange del primer dedo izquierdo de la mano, de 4x3x3 cm, sin afectación del segundo dedo. El tumor se encontró presente en tejido óseo y partes blandas, con necrosis focal e invasión vascular y perineural. Se observó tumor en todos los bordes de sección quirúrgica y tejido óseo distal.
Las técnicas de inmunohistoquímica informaron de tumor neuroendocrino primitivo Ewing/PNET. EMA (-), CKAE1 (-), Bcl-2 (-), S-100 (-), calretinina (-), CD34 (-), desmina (-), CD68 (-), CD56 (+), CD99 (+), VIM (+) focal, Ki67 (+) 80 %. (Figura 2).
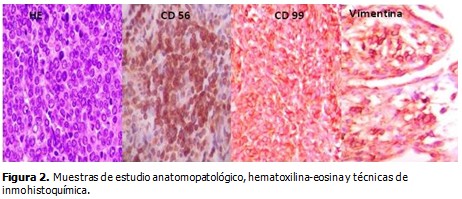
El 24 de marzo de 2015 (a un mes de la cirugía) fue valorada por medicina oncológica; fue replanteado el estadio, pT1 N0 M0 G3 etapa IIA, y se decidió comenzar con quimioterapia adyuvante esquema P-6 VAC-IE (bloque A con Ciclofosfamida, MESNA, Adriamicina y, Vincristina; y el bloque B con Ifosfamida, MESNA y Etopósido).
Se realizaron otros estudios de extensión como:
Biopsia de medula ósea: negativa.
Ecocardiograma: dentro de los parámetros normales con fracción de eyección de ventrículo izquierdo (FEVI): 60,9 %.
Gammagrafía ósea: negativa.
Hematología y hemoquímica dentro de los parámetros normales.
TAC de tórax, abdomen y pelvis: ausencia de lesiones pleropulmonares secundarias, hígado de tamaño normal sin imagen nodular definida, bazo, páncreas, riñones, vesícula, vías biliares y glándulas suprarrenales normales. Múltiples adenopatías intrabdominales e inguinales menores de 10mm, cambios osteodegenerativos y cambios posquirúrgicos en la mano izquierda.
Al concluir este informe, la paciente se encontraba recibiendo cuarto ciclo de quimioterapia esquema P6 bloque B, con buena tolerancia.
DISCUSIÓN
El sarcoma de Ewing pertenece a un grupo de tumores con aspectos histopatológicos y genéticos comunes, caracterizados por la presencia de translocaciones específicas [t(11;22) o t(21;22)] en el 95 % de los casos. Esta familia de tumores incluye al tumor de Askin (o PNET de la región toraco-pulmonar), el sarcoma de Ewing, PNET, PNET de hueso y sarcoma de Ewing extraóseo.2,6,7 Además, se caracterizan por la fuerte expresión de la glicoproteína MIC2 (CD99),8 y muestran también, de forma variable, tinción positiva para algunos marcadores neurales, como enolasa neuronal específica, sinaptofisina y S-100.2 Los resultados de los exámenes de inmunohistoquímica realizados a la paciente, coinciden con lo reportado en la literatura.6,7
La localización de mayor preferencia corresponde a las extremidades inferiores en el 41-45 % de los casos, seguida frecuentemente por los huesos de la pelvis del 20 al 26 %; luego están las extremidades superiores (9 %), columna vertebral (6 %), pies y manos (3 %) y cráneo (2 %).9,10 Estas cifras evidencian la localización poco usual del tumor en el caso presentado.
Entre un 20 y 25 % de pacientes son diagnosticados con enfermedad metastásica (10 % pulmón, 10 % hueso, 5 % médula ósea).11 Los estudios para establecer el estadio deben estar dirigidos a la detección de estas metástasis. Múltiples metástasis óseas confieren un pobre pronóstico en relación con la metástasis pleuropulmomar (<20 % comparado con 20 %–40 % de supervivencia a los cinco años). Otros factores asociados al pronóstico desfavorable, son el tamaño y el volumen tumoral, los niveles de LDL (lactato deshidrogenasa), la localización en el esqueleto axial, pacientes mayores de 15 años, una pobre respuesta histológica a la quimioterapia preoperatoria y la incompleta o la no realización de la cirugía.12
En el presente caso es necesario destacar que se hicieron modificaciones en el programa terapéutico. El tratamiento general para los tumores de la familia del sarcoma de Ewing, es la quimioterapia intercalada con cirugía, con o sin radioterapia en pacientes con enfermedad localizada. Deberá ser aplicada la radioterapia después de la cirugía, en casos con inadecuados márgenes quirúrgicos y se discutirá cuando la respuesta en el espécimen quirúrgico es pobre (más del 10 % de células tumorales viables).12 En esta paciente, luego del tratamiento quirúrgico se planificó quimioterapia esquema VAC/IE y luego radioterapia sobre el lecho quirúrgico.
El tratamiento primario consiste en quimioterapia con múltiples agentes. El esquema VAC/IE (Vincristina, Doxorrubicina y Ciclofosfamida alternado con Ifosfamida y Etopósido) es el régimen preferido para pacientes con enfermedad localizada, mientras que VAdriaC (Vincristina, Doxorrubicina y Ciclofosfamida) es el preferido para los pacientes con enfermedad metastásica. A los pacientes con enfermedad estable o mejoría después del tratamiento primario, se le puede realizar el tratamiento local, que incluye resección amplia, radioterapia definitiva con quimioterapia o, en casos seleccionados, amputación.13
Los sarcomas de Ewing/PNET son un grupo heterogéneo de tumores de origen neuroectodérmico que tienen una alta incidencia de recaída locorregional y metástasis a distancia, por lo que su pronóstico en la mayoría de los casos es desfavorable. Los pacientes que lo padezcan deben ser tratados en centros especializados y con experiencia para su adecuado manejo multidisciplinario.
