INTRODUCCIÓN
Los fibrosarcomas, aunque raros, son los sarcomas no rabdomiomatosos más comunes en la infancia, la niñez y la adolescencia (1-3). La incidencia por edades es similar a la observada en los rabdomiosarcomas y es más probable antes de los 5 años de edad y entre 10 a 15 años. Se encuentran con más frecuencia en las extremidades. El 70 % de los fibrosarcomas congénitos aparecen distalmente. No existen diferencias significativas en cuanto a su distribución con respecto al sexo. La mayor parte de los autores sugiere que el pronóstico es mejor mientras más joven es el niño (4). El grado de supervivencia para los fibrosarcomas no metastásicos de la infancia es mayor de un 90%. (5)
Los fibrosarcomas son tumores de células alargadas, caracterizados por un patrón entrelazado de células tumorales con una gran cantidad de estroma colágeno emergiendo de tejido fibroblástico. (2)
Los fibrosarcomas pueden ser difíciles, si no imposibles de distinguir de los tumores desmoldes o de las fibromatosis agresivas. Para propósitos prácticos, los desmoldes, las fibromatosis y los fibrosarcomas de bajo grado de malignidad pueden ser tomados como un espectro de neoplasias similares o muy cercanamente relacionadas. Como características importantes para establecer la malignidad se incluyen el pleomorfismo nuclear, el índice mitótico y la basofilia. (5-8)
Debe hacerse diagnóstico diferencial con rabdomiosarcomas indiferenciados, neurofibrosarcomas, fascitis nodular, miositis osificante y también con pseudotumores inflamatorios.
Los fibrosarcomas congénitos diagnosticados en edades tempranas están usualmente localizados de forma distal en las extremidades superiores o inferiores. La localización en el tronco es poco frecuente.
Se aprecia una masa dura, en ocasiones infiltrante, que puede estar fija a la piel o a tejidos profundos.
Los fibrosarcomas en la cabeza o en el cuello asociados a trastornos de la deglución o de la ventilación, pueden requerir de una traqueotomía previa a cualquier otro proceder. Las lesiones de alto grado de malignidad pueden metastizar tempranamente en hígado y pulmón o invadir estructuras óseas o nerviosas.
Algunas veces se pueden encontrar lesiones multifocales con múltiples tumores primarios afectando una extremidad. (3)
El tratamiento del fribrosarcoma es la resección quirúrgica con bordes de sección negativos de células neoplásicas, si esto puede ser realizado sin un significativo debilitamiento o deformidad de la extremidad. Ocasionalmente los pacientes se presentan con lesiones muy extensas o que progresan rápidamente, bajo estas circunstancias debe considerarse la posibilidad de quimioterapia sistémica preoperatoria para disminuir su tamaño. En caso de que la resección completa del tumor requiera de una intervención mutilante, como una amputación o la laringectomía, es preferible observar cómo se comporta el crecimiento del tumor, el cual puede permanecer sin crecimiento aparente permitiendo así el desarrollo normal del miembro afectado y la posibilidad de una resección menos deformante o debilitante. (4)
Las resecciones mutilantes deben realizarse solamente si el crecimiento del tumor es muy progresivo o si causa severos síntomas.
No está claramente definido el papel de la quimioterapia, porque ocasionalmente lesiones de alto grado de malignidad (especialmente en niños muy jóvenes) responden de diferente forma a una variedad de agentes. La quimioterapia debe ser utilizada en las lesiones progresivas, en las sintomáticas y en las irresecables.
En un estudio prospectivo randomizado se encontró que el uso de la radioterapia estuvo asociado con la disminución del grado de recurrencia en los sarcomas de baja malignidad con bordes de sección positivos , sin embargo, la mayor parte de los pacientes deben ser tratados de inicio con la escisión quirúrgica amplia.
PRESENTACIÓN DEL CASO
Se presenta el caso de la paciente OR, de la raza negra, sexo femenino, de 20 años de edad, de procedencia rural, con antecedentes de salud.
La paciente refirió que a la edad de 15 años comenzó a notar una “pelotita” en la cara interna del brazo izquierdo, de crecimiento lento pero progresivo.
Como procedía de un lejano lugar al norte del país, sin asistencia médica profesional acudió, como sus ancestros, al curandero de la región, quien le hizo ingerir diferentes brebajes y le practicó varias incisiones en la piel sobre el tumor, proceder que repitió en varias ocasiones sin que se lograra detener el crecimiento de este. Imposibilitada de realizar ninguna tarea con dicha extremidad, fue traída a la capital del país a solicitar atención médica en el Hospital “Princesa Marina” de Bostwana, donde fue atendida en la sección de Accidentes y Emergencias e ingresada en la sala de Cirugía, el 2 de Febrero de 2003.
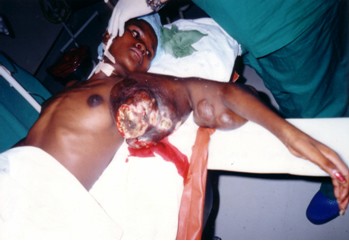
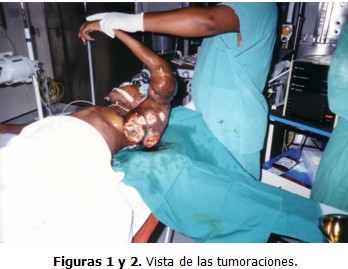
Al examen físico se apreciaba una gruesa tumoración (aproximadamente del tamaño de una cabeza humana) situada en la cara interna del brazo izquierdo y otras más pequeñas (como naranjas) agrupadas hacia el codo del mismo brazo, de consistencia dura, adheridas a la piel que las recubría, sin adherencia a planos profundos con múltiples áreas de necrosis, tejido esfacelado y sepsis localizada con gran fetidez. (Figuras 1 y 2)
Para caminar estaba obligada a apoyar el puño en la cadera y estaba imposibilitada de hacer cualquier movimiento con dicho brazo, ya que el peso y el tamaño del tumor obstaculizaban los normales movimientos del miembro.
Los exámenes complementarios revelaron los siguientes resultados:
Hemoglobina: 6,1 g/L.
Eritrosedimentación: 79 mm/h.
Ultrasonido abdominal: hígado aumentado de tamaño con ecopatrón normal; no imagen de metástasis hepática.
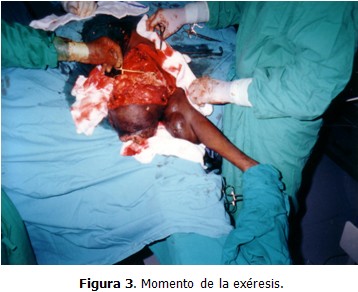
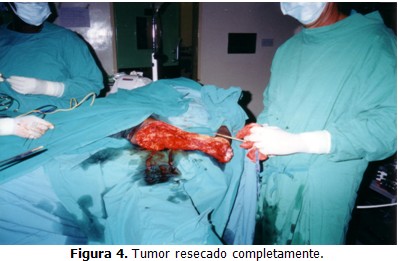
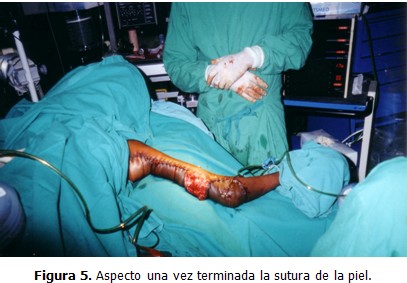
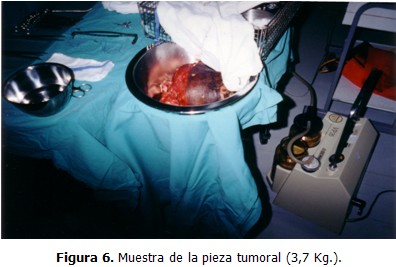
Los familiares no permitieron hacer biopsia para determinar el tipo histológico y abogaron por la operación lo antes posible, la cual fue efectuada el 10 de febrero de 2003. El tumor estaba encapsulado y fue posible su enucleación sin afectar el pedículo vásculonervioso del brazo, quedando sólo una pequeña área de la herida quirúrgica sin cubrir con la piel, la cual se planeaba injertar en un segundo tiempo para lograr el cierre por tercera intención. (Figuras 3, 4, 5 y 6)
La paciente evolucionó normalmente en sala durante una semana, luego comenzó a manifestar debilidad en las piernas y dificultades para caminar; la pérdida de la fuerza muscular iba acompañada de trastornos sensitivos, por lo que se realizó una tomografía axial computarizada de cráneo y columna vertebral, observándose múltiples metástasis en esta última.
La paciente dejó de levantarse dos semanas después de la operación, al perder la movilidad de las piernas. Comenzó a tener fiebre, tos y expectoración abundante, y a pesar del tratamiento antibiótico indicado, falleció a causa de bronconeumonía en marzo de 2003.
A causa de la no existencia de morgue en el hospital, fue imposible realizar la necropsia del cadáver, y dado el escaso número del personal técnico del Departamento de Anatomía Patológica, el informe de la biopsia de la pieza estuvo disponible en abril de 2003, concluyendo como sarcoma no rabdomiomatoso de partes blandas.
DISCUSION DEL CASO
La frecuencia de aparición de los sarcomas nunca rebasa el 1 al 1,5 % de todos los tumores malignos que se presentan, tanto en el hombre como en la mujer. (9-11)lo que evidencia su escasa frecuencia de presentación.
No se encontró ningún antecedente de presentación de un sarcoma no rabdomiomatoso de partes blandas en nuestra provincia, ni tampoco ninguna publicación de carácter nacional o internacional al respecto, en los sitios de salud a que tuvimos acceso.
Aún cuando en nuestro medio no se haya detectado ningún caso, consideramos que es de gran importancia el conocimiento de la existencia de esta entidad, por lo que puede aportar a la amplitud de la información al numeroso personal médico, que en todos las latitudes del mundo presta sus servicios, en algunas de cuyas regiones sí se detectan estos tumores, aunque por el deficiente sistema organizativo de salud, educación y superación no existan trabajos científicos serios al respecto.