INTRODUCCIÓN
Los medios de enseñanza, son parte importante e imprescindible en todo proceso de enseñanza, pues interactúan con los restantes componentes curriculares (objetivos, contenidos, estrategias, actividades, etc.), condicionando y modulando la prefiguración de los mismos, y viceversa. Por ello, no se concibe, en los momentos actuales, que un profesor desarrolle la docencia, empleando exclusivamente la palabra oral y sus gestos; incluso en las clases que pudiéramos considerar como más tradicionales, la pizarra y los libros son parte integrante y necesaria de las mismas.
El empleo en clases, de un libro o un vídeo, bien sobre la Edad Media, o bien sobre la fauna africana, propicia que los alumnos accedan a realidades que desde su marco vital, no pueden conocer. Es decir, los medios permiten obtener conocimiento, a través de experiencias de aprendizaje mediadas figurativa o simbólicamente; esto se incrementa las posibilidades de adquisición del conocimiento, más allá de la mera experiencia contingente o directa sobre la realidad circundante.
Actualmente, el material de estudio para nuestros educandos, es insuficiente, lo que desfavorece el correcto desenvolvimiento del proceso docente y exige la aplicación de variantes para resolver en alguna medida estas dificultades. En tal sentido, surge el interés por la realización de este folleto, el cual contiene información sobre las enfermedades metabólicas, estudiadas en la asignatura Metabolismo Intermediario y su Regulación que se imparten en el segundo semestre de primer año de la carrera de Medicina, como parte de la disciplina Bioquímica. Para su realización se realizó una búsqueda documental, mediante la técnica de revisión del contenido, en informaciones encontradas en Internet y en el fondo referativo de la biblioteca.
A través de este material de apoyo, se pretende de manera general, favorecer el desarrollo del proceso docente educativo, así como contar con un instrumento eficaz, para la preparación de los facilitadores en la aplicación del nuevo modelo pedagógico que actualmente se aplica..
Parte I: ALTERACIONES DEL METABOLISMO DE LOS GLÚCIDOS
1.1 Glucogenosis tipo I
La glucogenosis tipo Ia, también conocida como enfermedad de Von Gierke, déficit de glucosa-6-fosfatasa, glucogenosis hepatorrenal y depósito de glucógeno Ia, es una enfermedad metabólica rara, y de carácter hereditario, incluida dentro de los errores innatos del metabolismo. Pertenece al grupo de las glucogenosis, enfermedades producidas por depósito, o acumulación de glucógeno (sustancia que se forma en el organismo, para almacenar la energía que proviene de los hidratos de carbono). Está causada por un déficit congénito de la enzima glucosa-6-fosfatasa en hígado, riñón e intestino. Afecta la formación y utilización del glucógeno, dando lugar a concentraciones, o estructuras anormales del mismo.
El glucógeno es un polisacárido formado por moléculas de glucosa, unidas entre sí de una forma especial, que confiere a la molécula una estructura arbórea, que permite acumular millones de moléculas de glucosa. Se sintetiza y almacena en los tejidos hepático y muscular, y sus niveles pueden variar notablemente en ambos tejidos, como consecuencia de la alimentación y de los estímulos hormonales. En el hígado, su función es mantener la glucemia y alcanza una concentración de 70 mg/g de tejido, superior a la del músculo, 15 mg/g de tejido, dónde se utiliza para la obtención de energía, durante la contracción muscular.
El glucógeno se sintetiza en el tejido hepático, fundamentalmente a partir de la glucosa. Una vez dentro de los tejidos, la glucosa se transforma en glucógeno mediante una cadena de reacciones enzimáticas, luego, mediante una hexocinasa, se transforma en glucosa-6-fosfato, que a su vez se convierte en glucosa-1-fosfato, a través de otra enzima: la fosfoglucomutasa. La glucosa-1-fosfato se transforma en uridindifosfato glucosa y posteriormente, se van añadiendo restos de glucosa, por acción de la glucógeno sintetasa. Finalmente, mediante la enzima ramificante se completa la estructura normal del glucógeno.
La degradación del glucógeno se lleva a cabo mediante dos sistemas enzimáticos: la fosforilasa y la enzima desramificante. La fosforilasa libera glucosa-1-fosfato. La enzima desramificante es una proteína bifuncional; su actuación incluye dos pasos: en el primero, deja un único resto de glucosa unido a la cadena central, en el segundo paso la degrada a glucosa libre. Esta degradación del glucógeno, se traduce en la formación de glucosa libre en un 8-10 % y de glucosa-1-fosfato en un 90 %. La glucosa-1-fosfato es convertida en glucosa-6-fosfato por acción de la fosfoglucomutasa. Para poder ser liberada al torrente sanguíneo y de este modo mantener la glucemia, la glucosa-6-fosfato debe ser desfosfatada a glucosa mediante la enzima glucosa-6-fosfatasa.
En el músculo, la glucosa-1-fosfato y la glucosa-6-fosfato, entran en la glucólisis para la obtención de adenosín trifosfato (ATP) durante la contracción muscular. La regulación del metabolismo del glucógeno en el hígado, se produce a través de la concentración de glucosa extracelular; el hígado puede dar o recibir glucosa, para mantener la glucemia, dependiendo de los niveles de glucosa extracelulares. En este mecanismo de regulación, las enzimas fosforilasa y sintetasa son las más importantes.
Hormonas como el glucagón, activan la glucogenólisis a través de una serie de reacciones en cascada, que utilizan el adenosín monofosfato (AMPc) para la activación de la fosforilasa y la inhibición de la sintetasa; por su parte, la insulina activa la síntesis de glucógeno.
Las glucogenosis pueden clasificarse en diferentes categorías, en función de su mecanismo fisiopatológico o de producción, según los defectos enzimáticos identificados y en función de características clínicas diferenciadas:
1. Glucogenosis de fisiopatología hepática hipoglucémica: incluye las glucogenosis tipo Ia, Ib, III y VI.
2. Glucogenosis de fisiopatología muscular: incluye las glucogenosis tipo V, VII y los defectos de la glucólisis que no causan acumulación de glucógeno.
3. Glucogenosis de fisiopatología peculiar: como las glucogenosis tipo II y IV.
En cuanto a la nomenclatura, suelen nombrarse indistintamente siguiendo la numeración romana, con el nombre del defecto enzimático, o utilizando el nombre propio.
En conjunto, la prevalencia de las glucogenosis es de 1 por cada 20 000-25 000 nacidos vivos, con mayor frecuencia para los tipos I, II, III y IV. Todas se heredan como un rasgo genético autosómico recesivo, con excepción de la deficiencia de fosforilasa-b-cinasa, que está asociada al cromosoma X.
Esta enfermedad fue descrita inicialmente por Simón Van Creveld y por Edgar Von Gierke, a principios de los años treinta, con una frecuencia de 1/100 000- 400 000 habitantes. El primer caso diagnosticado de glucogenosis tipo I, es el publicado por Van Crefeld en 1928, y estudiado histológicamente por Von Gierke en 1929.
Como se planteó anteriormente, la glucogenosis Ia es una enfermedad que se hereda en forma autosómica recesiva, lo que significa que para transmitirla al hijo, tienen que ser portadores de esa información genética, tanto el padre como la madre. Por tanto, en estos casos las probabilidades de tener un hijo sano serían de un 25 %, las de tener un hijo portador de la enfermedad pero que no la desarrolle de un 50 % y las de tener un hijo enfermo 25 %. Existen casos de padres con tres hijos, de los cuales los dos primeros son sanos y el tercero padece esta enfermedad, pero también son frecuentes los casos en los que dos o tres hijos han desarrollado la enfermedad. En la actualidad, se han localizado las mutaciones genéticas que originan esta enfermedad, lo cual ha favorecido el estudio pre y posnatal y disminuye la recurrencia a técnicas invasivas como la biopsia hepática para la diagnosis enzimática.
Brody et al. identificaron en 1995, el gen de la glucosa-6-fosfatasa en el cromosoma 17q21, cuya reproducción reciente y la identificación de varias mutaciones causantes de la enfermedad, han mostrado una heterogeneidad molecular étnica, es decir, las mutaciones genéticas más frecuentes en los asiáticos o los chinos, no son las mismas que las encontradas por ejemplo, en los hispanos. Un estudio de Lei et al. en el mismo año, mostró que R83C y Q347X, eran las mutaciones más frecuentes en los caucasianos, R83C y 130X eran las más frecuentes en hispanos y R83H la más frecuente en chinos.
La actividad de la enzima glucosa-6-fosfatasa, puede conservarse en un pequeño porcentaje, o estar anulada, lo que depende de las mutaciones que provoquen su deficiencia. En un estudio realizado en el Centro Médico de Soroka (Israel), se observó que la mutación E110Q conservaba un 17 % de la actividad enzimática de la glucosa-6-fosfatasa, mientras que la D38V la anulaba.
Manifestaciones clínicas
Clínicamente se manifiesta a partir del primer año de vida y se caracteriza por aumento del tamaño del hígado y los riñones, cara redondeada (“de muñeca”), detención del crecimiento, desarrollo mental normal y ocasionalmente por hipotonía (tono anormalmente disminuido del músculo) leve. Además, puede existir una tendencia a la hipoglucemia (niveles bajos de glucosa en sangre) que se tolera bien, aunque sea muy severa, neutropenia (niveles anormalmente bajos de neutrófilos, un tipo de células blancas de la sangre), hiperlipemia (aumento de los lípidos en sangre, en general aumento del colesterol y triglicéridos) con producción de xantomas (tumores cutáneos que contienen ésteres de colesterol), hiperacidemia (aumento de ácidos sanguíneos) úrica con gota y hemorragias frecuentes por deterioro secundario de la función plaquetaria.
Se produce acidosis láctica (estado metabólico en el que existen cantidades anormales de cuerpos cetónicos) recidivante (recidiva es la aparición de una enfermedad en un individuo que ya la ha padecido hace algún tiempo), que puede ser grave y poner en peligro la vida del paciente.
En niños pequeños, puede verse una gran hepatomegalia (hígado anormalmente grande), que puede ser poco llamativa en la vida adulta, en la
que aparece una esplenomegalia discreta, y son los riñones los que se agrandan, moderada pero constantemente. Los recién nacidos con esta enfermedad, suelen presentar hepatomegalia (barriga hinchada por agrandamiento del hígado, que a veces se confunde en esta temprana edad con problemas de gases), y en ocasiones, convulsiones por hipoglucemias. Gradualmente, van apareciendo otros síntomas, como vómitos frecuentes, somnolencia, poca actividad, inapetencia e infecciones respiratorias y del intestino (gastroenteritis). En algunos casos de crisis convulsiva prolongada, es probable la ocurrencia de apnea (el cerebro se queda sin oxígeno y el cuerpo adquiere rigidez y un color morado) e incluso pueden llegar al estado de coma. En estos casos, es muy importante evitar una bronco aspiración con su propia lengua y acudir inmediatamente a un hospital.
Diagnóstico
El diagnóstico definitivo se lleva a cabo mediante determinación de los niveles de la enzima glucosa-6-fosfato-fosfatasa y por la presencia de agregados de glucógeno en el tejido hepático.
Es necesario tener la precaución, de no efectuar pruebas de tolerancia con galactosa o fructosa, ya que no se convierten en glucosa, lo que puede desencadenar una acidosis muy grave. Está indicado realizar una prueba del glucagón en ayunas, cuando se sospeche esta enfermedad, pero sin que existan evidencias de hipoglucemia y de acidosis láctica.
El diagnóstico diferencial debe hacerse, obligatoriamente, con la glucogenosis Ib, en la cual la enzima deficitaria es la translocasa microsomal y la forma de presentación es similar, pero más grave (con neutropenia severa e infecciones recurrentes).
No es posible en la actualidad el diagnóstico prenatal.
Pronóstico
Puede ser regular o bueno, ya que a medida que el paciente crece, sus problemas metabólicos van siendo menos graves y más fácilmente controlables. La hiperuricemia, como consecuencia del aumento del metabolismo de las purinas y de la insuficiencia renal, adquiere más relevancia a partir de la adolescencia. Pueden desarrollarse adenomas (tumor benigno de estructura semejante a una glándula) hepáticos, que en algunos casos pueden tornarse malignos; aparecen diarreas por la deficiente absorción intestinal de la glucosa y osteoporosis por efecto crónico de las acidemias y la insuficiencia renal.
Cuando la enfermedad ha alcanzado su pleno desarrollo, se presenta una marcada hepatomegalia, niveles altos de triglicéridos, colesterol, ácido láctico y también es frecuente la elevación del ácido úrico y la aparición de hemorragias y xantomas.
Tratamiento
En 1974, se descubrió que si se mantenían los niveles normales de glucosa durante el día y la noche, la mejoría era considerable. Durante el día, la glucosa se mantenía a través de alimentaciones frecuentes y de noche, mediante la infusión continua de una solución concentrada de glucosa en el estómago.
Chen et al. en 1984, experimentaron con la dieta de maicena como sustituto de las infusiones continuas durante la noche, para evitar las hipoglucemias. Esta terapia resultó ineficaz en niños con niveles bajos de la actividad pancreática, no así en otros casos, en los que la frecuente administración oral de maicena cruda, al igual que una nutrición parenteral total, o bien, infusión nasogástrica nocturna de glucosa, invirtieron todas las muestras físicas y bioquímicas de la enfermedad.
Lee et al. en 1996, probaron que el uso de maicena cruda, mantenía una glucemia adecuada durante una media de 4,25 horas (rango entre 2,5 y 6 horas).
La maicena es difícil de absorber, por lo que durante su digestión, pequeñas cantidades de glucosa van pasando a la sangre y se mantiene por más tiempo la glucemia.
Estos pacientes necesitan aportes de vitaminas y calcio; sin embargo, el aporte de proteínas debe de manejarse cuidadosamente, cuyo exceso puede elevar el ácido úrico.
El tratamiento de elección para evitar la hipoglucemia y la acidosis láctica, es la ingestión de glúcidos cada 3 ó 4 horas, que en los niños debe administrarse por sonda nasogástrica durante la noche.
La dieta de los pacientes con esta enfermedad, debe contener un 60 % de hidratos de carbono, exentos de galactosa o fructosa. Se añaden restricciones dietéticas para controlar otras alteraciones metabólicas, solo cuando se hace necesario. En algunos pacientes también se está utilizando fécula de mandioca para mantener su glucemia.
En los casos en que los niveles de ácido úrico son altos, se indica el tratamiento con alopurinol. En Estados Unidos de América, algunos pacientes con hiperactividad y síndrome de deficiencia de atención, se han tratado con ritalin (es una droga: methylphenidate), que cuenta con buen número de detractores; en consecuencia, se han presentado últimamente como sustitutos para mejorar estos problemas, complejos que proporcionan las dosis adecuadas de aminoácidos, minerales y vitaminas para el organismo.
La cirugía consistente en una derivación porto-cava, para prevenir la acidosis láctica, es clínicamente desalentadora. El transplante hepático puede considerarse en los casos graves, en los que han fracasado todas las otras posibilidades terapéuticas y para evitar las complicaciones de malignización.
1.2. Galactosemia
La galactosemia es la incapacidad del organismo para utilizar (metabolizar) el azúcar simple o galactosa (que causa la acumulación de galactosa 1 fosfato), que alcanza altos niveles en el organismo y causa lesiones al hígado, el sistema nervioso central y otros sistemas corporales. Recibe otras denominaciones: deficiencia de galactosa-1-fosfatouridil transferansa, deficiencia de galactocinasa y deficiencia de galactosa-6-fosfato epimerasa. La galactosemia clásica es una enfermedad grave, con un inicio temprano de los síntomas. Es una enfermedad hereditaria, transmitida como un rasgo autosómico recesivo y cuya ocurrencia es aproximadamente de 1 por cada 60 000 nacimientos.
El recién nacido recibe normalmente, hasta un 20 % de su consumo calórico en forma de lactosa, formada por glucosa y galactosa. Sin las enzimas transferasa, el lactante no es capaz de metabolizar la galactosa 1 fosfato, cuya acumulación ocasiona una lesión de las células parenquimatosas del riñón, el hígado y el cerebro. Esta lesión puede iniciarse prenatalmente, como consecuencia de la galactosa que le llega transplacentariamente, procedente de la dieta de la madre (heterocigótica), capaz de metabolizar la galactosa aunque con una eficacia reducida.
Las personas con galactosemia son incapaces de descomponer completamente el azúcar simple galactosa, que compone la mitad de la lactosa, el azúcar que se encuentra en la leche. La lactosa es un disacárido, lo que significa que está compuesto por dos azúcares enlazados: galactosa y glucosa.
Si a un bebé con galactosemia se le da leche, los derivados de la galactosa se acumulan en su sistema, causando daño al hígado, al cerebro, a los riñones y a los ojos. Los individuos con galactosemia no toleran ninguna forma de leche (humana o animal) y deben vigilar cuidadosamente la ingestión de otros alimentos que contengan galactosa. La exposición a los productos lácteos, puede ocasionar daño hepático, retardo mental, formación de cataratas e insuficiencia renal.
Manifestaciones clínicas
Clínicamente puede manifestarse mediante ictericia, hepatomegalia, vómitos, hipoglucemia, convulsiones, letargia, irritabilidad, dificultades en la alimentación, escaso aumento de peso, aminoaciduria, cataratas que se deben al aumento del galactitol, cirrosis hepática, ascitis, esplenomegalia o retraso mental.
Los pacientes con galactosemia presentan un mayor riesgo de sepsis neonatal por Escherichia coli, cuya aparición precede, a menudo, al diagnóstico de galactosemia. Cuando el diagnóstico no se realiza al nacer, tanto la lesión hepática (cirrosis) como la cerebral (retraso mental), van adquiriendo mayor gravedad hasta llegar a ser irreversibles. (Figura 3)
Síntomas
Debe considerarse la posibilidad de una galactosemia, ante un recién nacido o lactante que no se desarrolle normalmente o que presente síntomas tales como: ictericia (coloración amarillenta de la piel y de la esclerótica), vómitos, alimentación deficiente (el bebé se niega a beber fórmula que contenga leche), poco aumento de peso, letargo, irritabilidad y convulsiones.
Diagnóstico
El diagnóstico preliminar de la galactosemia, se realiza demostrando la presencia de una sustancia reductora, en varias muestras de orina, recogida mientras el paciente es alimentado con leche materna, leche de vaca o leche artificial que contenga lactosa. La sustancia reductora hallada en la orina, puede identificarse mediante cromatografía, o con una prueba enzimática específica para la galactosa.
Pronóstico
El niño puede vivir una vida normal, si se hace un diagnóstico temprano y se evitan estrictamente los productos lácteos.
Complicaciones
Las complicaciones pueden ser diversas: cataratas, cirrosis, infección grave por bacterias (sepsis por Escherichia coli), retraso en el desarrollo del lenguaje, retardo mental severo, ciclos menstruales irregulares, disminución de la función de los ovarios que conduce a insuficiencia ovárica, e incluso la muerte.
Tratamiento
Algunos lactantes con galactosemia, son capaces de tolerar cantidades elevadas de alimento con contenido de lactosa, pero ello es muy poco frecuente. Generalmente, es preciso excluir la galactosa de su dieta, en una fase temprana de la vida.
Una vez que se ha diagnosticado la enfermedad, se realiza un tratamiento que consiste en la abstinencia estricta del consumo de todos los tipos de leche y los productos lácteos. El niño puede ser alimentado con fórmulas basadas en soja, carne o nutramigen (una fórmula procesada a base de proteína hidrolizada). La galactosemia es una condición de por vida, por lo que el paciente debe evitar el consumo de estos productos durante toda su existencia.
Prevención
El conocimiento de la historia familiar es muy importante. Si existen antecedentes familiares de galactosemia, la asesoría genética puede ayudar a los futuros padres a tomar una decisión sobre el embarazo y las pruebas prenatales. Una vez que se hace el diagnóstico de galactosemia, se recomienda la asesoría genética para otros miembros de la familia.
Muchos estados establecen un examen obligatorio para detectar la galactosemia en recién nacidos, que de resultar positivo, los padres deben suspender inmediatamente los productos lácteos y hacerse un examen de sangre para galactosemia, por medio de su médico.
1.3. Deficiencia de glucosa 6 fosfato deshidrogenasa
El déficit de la glucosa 6 fosfato deshidrogenasa (G-6-PD), se debe a la herencia de cualquiera de un gran número de alelos anormales, del gen que codifica su síntesis. Se han encontrado más de cien variantes de G-6-PD, asociadas con un amplio espectro de enfermedades hemolíticas. La síntesis de la enzima del eritrocito, está codificada por un gen localizado en el cromosoma X y por esta razón la enfermedad es más común en los varones.
En el tipo de deficiencia más frecuente, la hemólisis se hace evidente entre las 48-96 horas posteriores a la ingestión de alguna sustancia con propiedades oxidantes, entre ellas determinados fármacos como antipiréticos, antipalúdicos, sulfamidas, etc. El grado de hemólisis varía de acuerdo con el agente, la cantidad ingerida y el grado de deficiencia enzimática.
La función más importante de la vía de las pentosas, en el eritrocito, consiste en la producción del cofactor NADPH, que se utiliza en la reducción del glutatión. Esto es esencial para la inactivación de compuestos oxidantes. Si alguno de los componentes del sistema falla, y disminuye la concentración del glutatión, la hemoglobina puede precipitar y provocar alteraciones en la membrana del hematíe. Esta distorsión estimula la eliminación de los eritrocitos por el bazo, lo que puede dar lugar a un proceso hemolítico agudo. (Figura 4)
La deficiencia de G-6-PD es un defecto hereditario recesivo ligado al cromosoma X, cuyo efecto primario consiste en la disminución de la producción de la enzima G-6-PD en los glóbulos rojos. Esto produce hemólisis y anemia de tipo hemolítica aguda, o esferocítica crónica.
En los Estados Unidos, la incidencia de este trastorno es mucho más alta entre la población de raza negra, con una frecuencia heterocigótica (estado de portador, en la que un gen es normal y el otro gen es anormal) del 24 %. Aproximadamente entre el 10 y el 14 % de esa población, se ve afectada por este trastorno, que además incide con mayor severidad en los hombres que en las mujeres (en dependencia de su herencia genética). Las personas que sufren esta enfermedad, normalmente no son anémicas y son asintomáticas hasta el momento en que sus glóbulos rojos, son expuestos a una sustancia oxidante o al estrés. Entre los medicamentos que pueden precipitar la reacción, pueden citarse los siguientes: agentes antimaláricos, sulfonamidas (antibióticos), aspirina, medicamentos antiinflamatorios no esteroideos (AINES), nitrofurantoína, quinidina, quinina y otros. También puede originarse debido a la exposición a ciertas sustancias químicas, como las presentes en las bolas de naftalin. La anemia esferocítica crónica no es afectada por estos medicamentos. La búsqueda de antecedentes de anemias hemolíticas o esferocitosis, en la historia familiar, puede disminuir el riesgo de sufrir una crisis hemolítica; aunque también puede realizarse un examen para descartar esas condiciones médicas, antes de suministrar alguno de los medicamentos vistos anteriormente.
Los episodios generalmente son cortos, ya que los glóbulos rojos nuevos (jóvenes) tienen una actividad normal de la G-6-PD.
Los factores de riesgo para desarrollar esta enfermedad, son: ser de raza negra, del sexo masculino y tener antecedentes familiares de deficiencia de G-6-PD. Un tipo diferente de deficiencia de G-6-PD se presenta en personas de raza blanca con orígenes en la zona geográfica de la cuenca mediterránea, el cual también se asocia con episodios agudos de hemólisis, pero de mayor duración y severidad.
Síntomas
Pueden aparecer como síntomas: fatiga, palidez, dificultad respiratoria, frecuencia cardiaca rápida (taquicardia), coloración amarilla de la piel (ictericia), orina de color oscuro y agrandamiento del bazo.
Signos y exámenes
Los signos que pueden remitir a esta enfermedad, son la anemia, la hemólisis, la reducción de la actividad de la G-6-PD y la reticulocitosis después de una crisis hemolítica; mientras que en los exámenes de laboratorio se buscarán alteraciones como, bilirrubina elevada, haptoglobina sérica disminuida, hemoglobina en la orina (hemoglobinuria), conteo absoluto de reticulocitos elevado, conteo disminuido de glóbulos rojos y de hemoglobina y presencia de cuerpos de Heinz en el extendido de sangre periférica al usar tinciones especiales. También se realiza el examen del azul de metileno y el de la reducción de la metahemoglobina.
Tratamiento
Se debe tratar la causa que ha precipitado, o causado la crisis. Es decir, si la causa es una infección, esta debe ser tratada; si es un medicamento, este debe ser suspendido.
Es posible que las personas con la forma mediterránea de la enfermedad, o en general aquellos que presentan una crisis hemolítica, requieran transfusiones.
Pronóstico
Lo normal es la resolución espontánea de los síntomas de la crisis hemolítica, o sea, sin necesidad de tratamiento.
Complicaciones
En raras ocasiones se produce la muerte, a causa de un evento hemolítico severo.
Prevención
Las personas que padecen la deficiencia de G-6-PD, deben evitar estrictamente la exposición a los factores que pueden precipitar un episodio de agudización, especialmente a aquellos medicamentos que se sabe producen reacciones de oxidación.
Parte II: ALTERACIONES DEL METABOLISMO DE LOS LÍPIDOS
2.1. Obesidad
La obesidad es un problema crónico caracterizado por un exceso de grasa corporal, de elevada prevalencia y de demanda asistencial creciente. La obesidad puede definirse, como un exceso de grasa que condiciona la salud de la persona. Cuando la cantidad de energía que se ingiere con los alimentos, es superior a la que se gasta, el exceso de energías es transformado en grasas. La definición de obesidad es arbitraria, pero parece confirmado que los riesgos para la salud son significativos, cuando el sobrepeso alcanza el 20 o el 25 %.
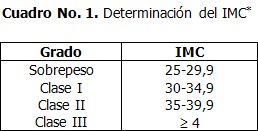
La medida más fácilmente realizable y mejor correlacionada con la grasa corporal total es el Índice de Masa Corporal (IMC), o Índice de Quetelet: peso/ talla 2. Aunque hay autores que sitúan el límite en 27,8 para los varones y 27,3 para las mujeres, la mayoría de los consensos definen la obesidad a partir de un IMC de 30. El IMC no es buen indicador en la niñez, la adolescencia, ni en ancianos, o en personas muy musculosas; aún así, es la medida más útil para hacer el seguimiento de la pérdida de peso y determinar la eficacia de un tratamiento. Igualmente, resulta más acertada la clasificación de la obesidad basada en el IMC. (Cuadro 1)
Para valorar la obesidad se necesita de indicadores objetivos, tales como:
• Porcentaje del peso actual con respecto al peso ideal. Clásicamente se utilizan las tablas peso y talla realizadas por la Metropolitan Life Insurance Company.
• Índice de masa corporal (IMC), calculado por medio de la fórmula: IMC = Peso (Kg) / [Talla (m)]2.
• Medición de los pliegues subcutáneos. El que mejor valora el exceso de grasa, es el pliegue tricipital (pliegue formado a nivel del músculo tríceps).
La obesidad es una enfermedad con repercusiones sistémicas, pues implica riesgos considerables para la salud tales como: disminución de la esperanza de vida como factor de riesgo independiente del riesgo cardiovascular (en discusión), factor de riesgo para el desarrollo de diabetes mellitus, accidentes vasculares encefálicos (AVE), hipertensión arterial, hiperlipemia y cáncer (colon, recto y próstata en el varón; vesícula, ovario, mama, cervix y endometrio en la mujer). También determina una mayor probabilidad para complicaciones como la litiasis biliar, la esteatosis hepática, las irregularidades menstruales, la gota, la artrosis, las várices, las tromboembolias y la hernia de hiato. Se pueden diferenciar dos tipos de obesidad: obesidad primaria o de causa no delimitada y obesidad secundaria o de causa conocida. Una persona puede desarrollar un cuadro de obesidad por:
• Disminución del gasto energético (descenso en la energía que se consume).
• La presencia conjunta de los dos hechos mencionados.
• Aumento de la ingesta de energía.
El gasto energético que tiene una persona, depende de:
• Gasto energético basal: cantidad de energía que requiere el cuerpo
• Ayuno de 12 horas en estado de reposo y condiciones ambientales neutras.
• Termogénesis: gasto energético debido a los diferentes estímulos ambientales (calor, frío, etc.).
• Gasto energético que condiciona la actividad física: consumo de energía causado por las actividades diarias y el ejercicio que realiza el individuo.
Según la distribución de la grasa, se puede hablar de:
• Obesidad androide: También conocida como obesidad abdominal, se presenta en un mayor número de casos en varones, que en mujeres. Se caracteriza por la acumulación de grasas, por encima de la cintura. Es un factor predisponente para enfermedades como: hipertensión arterial, enfermedades cardiovasculares, colelitiasis, hiperinsulinismo y diabetes mellitus.
• Obesidad ginecoide: Es más frecuente en mujeres que en varones. Se caracteriza por la acumulación de grasa en el bajo vientre, caderas y muslos.
Causas
Las causas de obesidad son múltiples y poco conocidas en la actualidad, lo que explica su alta incidencia; pueden señalarse las siguientes:
• Aporte energético de la dieta. En los animales de experimentación, se ha demostrado que la hiperfagia (comer mucho) es una causa de obesidad, aunque actualmente, se discute si este hecho es causa o efecto.
• Neuroendocrinología. Siempre se pensó que la obesidad era una enfermedad endocrinológica. Actualmente se conoce que sólo el 3 % de los casos de obesidad, responden a este tipo de alteraciones. Entre las alteraciones endocrinológicas, que generalmente se asocian a la obesidad, están las hipotalámicas, las hipofisiarias, las suprarrenales, el hipotiroidismo grave y el síndrome de ovario poliquístico.
• Factores genéticos y ambientales. Incluyen los genes compartidos, la misma dieta y el mismo nivel cultural, así como otros factores relativos al estilo de vida
En ciertos casos, la obesidad se debe a alguna causa identificable, como el uso de determinados fármacos (antidepresivos, fenotiazinas, esteroides, antidiabéticos orales: sulfonilureas y meglitinidas), o el padecimiento de enfermedades específicas (hipotiroidismo, síndrome de Cushing, insulinoma y poliquistosis ovárica); es por esto, que se buscarán estas causas, ante una persona con un IMC >=30, para determinar si la obesidad es de causa secundaria.
Signos y síntomas
Los signos y síntomas más comunes, son el exceso de peso y el incremento de la grasa corporal; mientras que otros como la vida sedentaria y los problemas psicológicos (depresión, baja autoestima, etc.), derivan de otras afecciones asociadas a la obesidad.
Diagnóstico
Se investigarán las circunstancias de comienzo de la obesidad, incidiendo sobre todo en la fecha aproximada y en la concurrencia de factores como: embarazo, lactancia, matrimonio, desajuste afectivo, sedentarismo, cambios vitales importantes, etc. En general, las obesidades de larga evolución, en mujeres, con ganancia progresiva desde la adolescencia, tienen más riesgo. Una ganancia rápida en el último año, obliga a investigar cambios dietéticos recientes, secundarismo, o adversidades psíquicas.
De forma rutinaria, debe evaluarse el riesgo cardiovascular global, bien mediante cuantificación con tablas, o bien identificando los factores de riesgo cardiovascular asociados (diabetes, hiperlipemia, tabaquismo, hipertensión arterial). También se realizarán las pruebas adecuadas, si se sospecha secundarismo, o enfermedades relacionadas con la obesidad (función hepática, ácido úrico).
Tratamiento
El tratamiento puede ser dietético (dieta hipocalórica), quirúrgico, farmacológico o bien encaminado a cambios en el estilo de vida; este último requiere de atención psicoterapéurica, junto a cambios en el hábito alimenticio y la práctica de ejercicios.
Prevención
La prevención de la obesidad exige, principalmente, un estilo de vida adecuado que incluye no solo la ingestión de una dieta balanceada, capaz de aportar todos los requerimientos del organismo y en la que el aporte de energía se corresponda con el consumo, sino también la realización sistemática de ejercicios físicos.
2.2. Aterosclerosis
La aterosclerosis se caracteriza por depósitos de grasa y engrosamiento de la túnica íntima, con rotura de la media, en las arterias mayores y media. Es una combinación variable de cambios en la íntima, que incluye acumulación focal de moléculas-lípidos complejos, proteínas y carbohidratos, sangre con todos sus constituyentes y proliferación celular, acompañada por formación de tejido fibroso, calcificación y cambios asociados en la media, con deposición significativa de lípidos en la pared arterial, lo que reduce la elasticidad de las arterias y contribuye a la oclusión. (Figura 3)
El término aterosclerosis generalmente designa varias enfermedades, en las que se produce engrosamiento y pérdida de elasticidad de la pared arterial. En esta enfermedad, la acumulación de la materia grasa debajo del revestimiento interno de la pared arterial, afecta a las arterias del cerebro, el corazón, los riñones, otros órganos vitales, así como los brazos y las piernas. Cuando la aterosclerosis se desarrolla en las arterias que alimentan el cerebro (arterias carótidas), se puede producir un ictus; cuando se desarrolla en las arterias que alimentan el corazón (arterias coronarias), se puede producir un infarto de miocardio. En la mayoría de los países occidentales, la aterosclerosis es la enfermedad más frecuente y la causa principal de muerte, y representa el doble de las muertes por cáncer y diez veces más que por accidentes. A pesar de los significativos avances médicos, la enfermedad de las arterias coronarias y el ictus aterosclerótico, son responsables de más fallecimientos que las demás causas en su totalidad. Es además una afección multifactorial, que tiene como factores de riesgo la edad, el sexo masculino, la hipertensión arterial, la hiperlipidemia, el tabaquismo, la diabetes, la obesidad, el sedentarismo y los rasgos de la personalidad.
Causas
La aterosclerosis se inicia cuando unos glóbulos blancos llamados monocitos, emigran desde el flujo sanguíneo hacia el interior de la pared arterial y se transforman en células que acumulan materias grasas. Con el tiempo, estos monocitos cargados de grasa, se acumulan y producen engrosamientos irregularmente repartidos por el revestimiento interno de la arteria. Cada zona de engrosamiento (llamada placa aterosclerótica o ateroma), se llena de una sustancia blanda parecida al queso, formada por diversas materias grasas, principalmente colesterol, células musculares lisas y células del tejido conjuntivo. Los ateromas pueden localizarse en cualquier arteria de tamaño grande o mediano, pero, por lo general, se forman donde las arterias se ramifican (presumiblemente por la turbulencia constante de estas zonas, que lesiona la pared arterial).
Las arterias afectadas por la aterosclerosis pierden su elasticidad y, a medida que los ateromas crecen, se hacen más estrechas. Además, con el tiempo los ateromas acumulan depósitos de calcio, que pueden volverse frágiles y romperse. Por esta razón, la sangre puede entrar en un ateroma roto, aumentando su tamaño y disminuyendo aún más la luz arterial. Puede suceder también, que un ateroma roto derrame su contenido graso, lo que desencadena la formación de un coágulo sanguíneo (trombo), el cual provoca mayor estrechez de la arteria, e incluso puede ocluirla, o bien puede desprenderse y pasar a la sangre, hasta llegar a una arteria más pequeña, donde causará igualmente, una oclusión (embolia).
Análisis del componente celular
Uno de los primeros eventos en la génesis de la aterosclerosis, es la adhesión de monocitos circulantes a la superficie intacta de las células endoteliales.
Esto va precedido de la expresión de moléculas de adhesión a la célula vascular (VCAM: del inglés, vascular cell adhesión molecule), en determinadas partes del sistema circulatorio, como consecuencia de la inducción, por colesterol y otros lípidos, del gen de la VCAM. Las fuerzas de cizallamiento se producen por el paso de los elementos formes de la sangre, principalmente el eritrocito, a través de los vasos. Cuando ocurren fuerzas de cizallamiento anormales sobre la superficie de las células endoteliales, como suele suceder en la bifurcación de las arterias coronarias, pueden ser inducidos los genes cuyos productos contribuyen al desarrollo de la aterosclerosis. La célula endotelial funciona como un sensor, su superficie es capaz de detectar las alteraciones en el flujo sanguíneo y transmitir estas alteraciones al núcleo.
El monocito penetra hasta la íntima, donde es transformado en macrófago, el cual fagocita lípidos modificados. Dentro de los lisosomas secundarios de los macrófagos, se forman capas trilaminares que impiden el intercambio hacia el centro, por lo que el colesterol queda incomunicado y se cristaliza. Una vez formados los cristales de colesterol, desaparece la capa trilaminar, pero el macrófago ya se ha transformado irreversiblemente en célula espumosa, uno de los constituyentes primarios de la placa de grasa.
Mientras ocurre este proceso, células del músculo liso migran desde la media, hasta la íntima, donde al dividirse producen colágeno y otras moléculas de la matriz también se cargan de lípidos, por lo que contribuyen al volumen de la lesión.
Algunos autores plantean la teoría monoclonal, para explicar la proliferación de las células musculares lisas, pues en un individuo todas son del mismo tipo, lo que sugiere que deriven de una sola célula, que fue la que inicialmente migró.
También están involucradas las células T, que son linfocitos especializados activados al reconocer determinados antígenos, en la superficie de células potencialmente patogénicas y propician la destrucción de estas últimas al activar a los macrófagos.
Análisis de los factores moleculares
Entre los principales factores moleculares, se encuentran los factores de crecimiento, los eicosanoides, las citokinas y el óxido nítrico.
Los factores de crecimiento son aquellos que atraen a las células y promueven la división celular. Los eicosanoides, al estimular la hidrólisis de los ésteres de colesterol, producen colesterol libre. Las citokinas tienen varios efectos metabólicos, incluyendo la manifestación de factores que regulan la formación del coágulo sanguíneo. Por su parte, el óxido nítrico actúa dilatando los vasos sanguíneos.
La migración de las células del músculo liso, la proliferación, la síntesis de moléculas de la matriz y el secuestro de lípidos, están influidos por los factores de crecimiento y las citokinas producidas por los macrófagos, las células T y las células endoteliales.
Las células endoteliales y las plaquetas, producen el factor de crecimiento derivado de las plaquetas, que atrae a las células musculares lisas hacia la íntima y activan su proliferación.
Los macrófagos liberan citokinas y factores de crecimiento, que afectan la distribución de colesterol en el organismo y pueden incidir en la proliferación de las células del músculo liso. Las citokinas liberadas por los macrófagos, estimulan a las células endoteliales a expresar el factor de activación plaquetario, el factor hístico y el inhibidor del activador del plasminógeno; participan en la transformación de la superficie de la célula endotelial, de forma tal que favorecen la circulación sanguínea. Al mismo tiempo, la célula endotelial activa la liberación de un número de sustancias como la prostaciclina y el óxido nítrico, que impiden la formación del coágulo sanguíneo previniendo la agregación plaquetaria; pero además, provocan que las células del músculo de la arteria se relajen y que como respuesta a las fuerzas de cizallamiento, las células endoteliales promuevan la producción de citokinas y ciertos factores de crecimiento con actividad aterogénica.
Como ya se ha dicho, la aterosclerosis comienza cuando los monocitos que se hallan en la circulación sanguínea, entran en la pared arterial y se transforman en células que acumulan materias grasas, lo que provoca un engrosamiento en algunas zonas (placas) del revestimiento interno de la pared arterial. (Cuadro 2)
Colesterol y enfermedad coronaria
Existe una elevada incidencia de enfermedades coronarias en pacientes con elevados niveles de LDLc y colesterol total, cuya disminución se obtiene si se cumple con la dieta y se insertan los cambios necesarios en el estilo de vida; en ocasiones, es necesario adicionar el uso de hipolipemiantes.
En general, una disminución de 100 mg en el colesterol de los alimentos causa una reducción aproximada de 0,13 mmol.L en el suero.
Se considera que una reducción del 1 % en las cifras de colesterol total, puede conducir al 2 % de reducción del riesgo coronario.
Síntomas
Por lo general, la aterosclerosis no produce síntomas hasta que no estrecha gravemente la arteria, o causa una obstrucción súbita. Los síntomas dependen del lugar donde se desarrolla la aterosclerosis: el corazón, el cerebro, las piernas, o casi en cualquier parte del organismo.
El primer síntoma del estrechamiento de una arteria, puede ser un dolor o un calambre en los momentos en que el flujo de sangre es insuficiente para satisfacer las necesidades de oxígeno; por ejemplo: durante el ejercicio, una persona puede sentir dolor de pecho (angina), debido a la falta de oxígeno en el corazón; o mientras camina, pueden aparecer calambres en las piernas (claudicación intermitente), debido a la falta de oxígeno en las extremidades. Estos síntomas se desarrollan gradualmente, a medida que el ateroma constriñe la arteria. Sin embargo, cuando se produce una obstrucción súbita, los síntomas aparecen inmediatamente; por ejemplo: cuando un coágulo sanguíneo se enclava en una arteria.
Factores de riesgo
El riesgo de desarrollar aterosclerosis aumenta con la hipertensión arterial, los altos valores de colesterol, el tabaquismo, la diabetes, la obesidad, la falta de ejercicio y la edad avanzada. Tener un pariente cercano que ya ha desarrollado aterosclerosis a una edad temprana, también aumenta el riesgo. Los varones tienen un riesgo mayor de padecer esta enfermedad que las mujeres, aunque después de la menopausia, el riesgo aumenta en las mujeres y finalmente se iguala al de los varones.
Las personas con homocistinuria, una enfermedad hereditaria, desarrollan ateromas con gran facilidad, sobre todo en edad juvenil.
La enfermedad afecta a muchas arterias, pero no a las arterias coronarias que alimentan el corazón. Por el contrario, en la hipercolesterolemia familiar hereditaria, los valores extremadamente elevados de colesterol en la sangre, provocan la formación de ateromas en las arterias coronarias, mucho más que en las otras arterias.
Prevención
Para prevenir la aterosclerosis, se deben eliminar los factores de riesgo controlables, como los valores elevados de colesterol en la sangre, la presión arterial alta, el consumo de tabaco, la obesidad y la falta de ejercicio.
El hábito de fumar es particularmente peligroso para las personas que ya tienen un riesgo elevado de sufrir enfermedades cardiacas. Fumar cigarrillos disminuye la concentración del colesterol “bueno”, o colesterol con lipoproteínas de alta densidad (HDL) y aumenta la concentración del colesterol “malo”, o colesterol con lipoproteínas de baja densidad (LDL). Además, aumenta la tendencia de la sangre a coagularse, lo que incrementa el riesgo de enfermedad arterial periférica, enfermedad de las arterias coronarias, ictus y obstrucción de un injerto arterial tras una intervención quirúrgica.
El riesgo que tiene un fumador de desarrollar una enfermedad de las arterias coronarias, está directamente relacionado con la cantidad de cigarrillos que fuma a diario. Las personas que dejan de fumar, tienen la mitad del riesgo de los que siguen fumando (con independencia de cuánto hayan fumado antes de abandonar el hábito). Dejar de fumar también disminuye el riesgo de muerte tras una cirugía de revascularización coronaria o de un infarto. También disminuye la incidencia de enfermedades en general, así como el riesgo de muerte en pacientes con aterosclerosis en arterias que no alimentan el corazón y el cerebro.
Tratamiento
El mejor tratamiento para la aterosclerosis es la prevención. Cuando la aterosclerosis se vuelve lo suficientemente grave como para causar complicaciones, se deben tratar las complicaciones mismas (angina de pecho, infarto, arritmias, insuficiencia cardiaca, insuficiencia renal, ictus u obstrucción de las arterias periféricas). En el caso de los pacientes hipercolesteronémicos, la dieta hipolipemiante es el pilar de la terapia, al cual se adiciona el control del peso, la realización de actividad física regular y la eliminación del tabaquismo, que se resumen en la práctica de un estilo de vida adecuado.
La dieta hipocolesteromiante tiene como objetivos:
1. Reducir la grasa total a menos del 30 % del consumo total de energía.
2. Reducir las grasas saturadas a menos del 10 % del consumo total de energía.
3. Incrementar moderadamente el uso de aceites y grasas monoinsaturadas entre 10 y 15 % del consumo de energía total y poliinsaturadas entre 7 y 10 %.
4. Reducir el consumo de colesterol a menos de 300 mg/día.
5. Incrementar el consumo de carbohidratos complejos y fibra.
6. Seleccionar fuentes de proteína que a la vez sean bajas en grasas saturadas. Las proteínas deben representar aproximadamente un 15 % del consumo total de energía.
Para lograr los dos primeros, es importante evitar la ingestión de mantequilla, leche entera, crema de leche, helados, queso, carne de cerdo, embutidos y aceite de coco. Para lo tercero, habría que utilizar aceite para cocinar, en lugar de grasa animal. Para el cuarto objetivo, es favorable limitar el consumo de huevos (menos de tres por semana) y vísceras (solo una vez a la semana) y no consumir alimentos con chocolate. En cuanto al consumo de los carbohidratos complejos y fibra, ayuda el consumo de frutas y vegetales, excepto aguacate los granos secos y los cereales. En el caso de las proteínas, las más factibles serían pescado, pollo o pavo, desprovistos de piel, así como carnero. Además, se reduce el riesgo global si se regulan el consumo diario de sodio (menos de 2 400 mg/día) y de alcohol (menos de30 g/día).
2.3. Enfermedad de Tay Sachs
Se produce por depósito en los lisosomas y afecta fundamentalmente al sistema nervioso central, por lo que no se aprecian signos de depósito periférico manifiesto en la exploración física. Esta enfermedad es la más devastadora de todas las de su tipo y se da con frecuencia en los individuos de origen judío asquenazí.
Etiología
El defecto básico es un déficit de la enzima lisosómica termolábil beta hexosaminidasa A y B, que son responsables de la actividad total. Son necesarias dos cadenas polipeptídicas alfa y beta, para la formación de la beta hexosaminidasa A y B. Un defecto en la cadena beta afecta a la actividad de las dos isoenzimas A y B, y da lugar por tanto, a un déficit de la actividad total de la beta hexosaminidasa, la cual requiere para su actividad hidrolítica, un activador que se une a la enzima y al sustrato natural: el gangliósido GM2.
Manifestaciones clínicas
Los recién nacidos afectados, presentan un desarrollo normal hasta los cinco meses de edad aproximadamente. Por lo general, se observa primero una reducción del contacto ocular y del enfoque visual, junto con una respuesta de alarma excesiva a los ruidos (hiperacusia). Al final del primer año de vida, el niño con una enfermedad de este tipo presenta una hipotonía grave. La exploración física muestra, a menudo, hipotonía intensa, ceguera e hiperacusia. El examen del fondo de ojo puede poner de manifiesto una mancha de color rojo cereza en la mácula. Estos niños adoptan una postura de rana y tienen una interacción muy limitada con su entorno. El tamaño craneal puede aumentar en más de un 50 %, pero este agrandamiento no se asocia con una hidrocefalia. Pueden aparecer complicaciones como crisis convulsivas, en el segundo año de vida y generalmente fallecen entre los dos y los cuatro años.
Diagnóstico
La enfermedad de Tay Sachs se sospecha generalmente en niños pequeños, con retraso grave, con una mancha color rojo cereza en la mácula y con carencia de depósito visceral. Varias esfingolipidosis se asocian con una mancha de color rojo cereza, pero es posible distinguir entre una y otra porque los pacientes con la enfermedad de Tay Sachs, carecen de hepatoesplenomegalia. En edades juveniles, se sospecha en un niño en el que la ataxia y la disartria adquieren un carácter progresivo.
El ensayo de beta hexosaminidasa A es diagnóstico y puede efectuarse en plasma, fibroblastos cutáneos en cultivo, o leucocitos hemáticos. Los portadores de la enfermedad de Tay Sachs, pueden detectarse mediante un ensayo de la actividad específica de la hexosamina.
Tratamiento
No existe tratamiento alguno para ninguna de las formas de enfermedad de Tay Sachs.
Parte III: ALTERACIONES DEL METABOLISMO DE LOS COMPUESTOS NITROGENADOS
3.1. Alteraciones del metabolismo de los aminoácidos. Fenilcetonuria.
La fenilcetonuria es también conocida como déficit de fenilalanín hidroxilasa, fenilalaninemia, oligofrenia fenilpirúvica, Síndrome de Fölling, e hiperfenilalaninemia. Las hiperfenilalaninemias (HPA) constituyen un grupo de alteraciones en el metabolismo de la fenilalanina (FA), que presentan heterogeneidad genética, clínica y bioquímica, definidos como la elevación de FA en plasma por encima de 120 mmol/L (2 mg/dL).
Las primeras evidencias en relación con estas alteraciones, derivan de los estudios del científico noruego Asbjorn Fölling, que en 1934 reportó un grupo de pacientes retrasados mentales que excretaban fenilpiruvato en la orina, con olor característico, y encontró que estos mostraban además, altos niveles de FA en sangre. El defecto metabólico fue descubierto por Jervis, en 1947, quien planteó que el bloqueo estaba en la hidroxilación de la FA (conversión de la FA en tirosina), aunque se conocía muy poco del sistema enzimático responsable de ello.
El primer avance se produjo cuando se estableció que, como mínimo, dos proteínas intervenían en la hidroxilación. En 1957, Mitoma y Wallace reportaron que en el hígado de pacientes con fenilcetonuria (FCU) sólo estaba ausente la fenilalanina hidroxilasa (FAH) lábil, mientras que la estable, dihidrobiopterina reductasa (DHBR), estaba presente. Actualmente, se sabe que esta vía metabólica es un sistema complejo.
Una de las formas clínicas de HPA descrita es la benigna, con una mayor actividad enzimática, y la FA plasmática entre 240 y 600 µmol/L (4 a 10 mg/dL), que no requiere tratamiento dietético. Estas concentraciones no originan la excreción urinaria de ácido fenilpirúvico; para que esto ocurra la fenilalanina en plasma debe sobrepasar los 900 µmol/L (15 mg/dL). Estos individuos conservan cierta actividad enzimática residual, con cifras obtenidas que oscilan entre 10 y 35 % de lo normal, a diferencia de lo que ocurre en la FCU clásica, donde se observa una ausencia completa de la actividad de dicha enzima. Desde que se puso en práctica en Cuba el Programa de Detección Precoz de la FCU, se han hallado 56 niños con HPA benigna, a los que no se les ha realizado un seguimiento estrecho de la fenilalanina en plasma, ni de la presencia o no de signos y/o síntomas que puedan tener repercusión en su coeficiente de inteligencia (CI).
Las hiperfenilalaninemias (HFA) se producen por mutaciones en el gen que codifica para la enzima fenilalanina hidroxilasa, que actúa sobre la conversión de fenilalanina a tirosina; como consecuencia se acumula la fenilalanina en el organismo y se produce, como signo clínico principal, retardo mental, cuyo grado va a estar de acuerdo con la mutación, que es la que define que la enzima tenga más o menos actividad.
Cuando los casos son más severos se conocen como fenilcetonuria. Se pueden diagnosticar mediante pruebas que detecten la presencia de fenilalanina en la sangre, como el método microbiológico de Guthrie, que consiste en la toma de sangre capilar de los niños al nacer, para compararla el nivel de crecimiento inducido por la muestra con los niveles estándar, en una cepa de la bacteria bacillus subtilis que requiere fenilalanina para su crecimiento. El pesquisaje neonatal es considerado el mejor método para la detección temprana de las HFA.
En Cuba, a partir de 1984, como parte del programa de diagnóstico y prevención de enfermedades hereditarias, comienza a realizarse el pesquisaje de FCU, lo que ha permitido que todos los casos detectados se encuentren bajo tratamiento dietético. En muchos países desarrollados, el tratamiento se realiza de forma más individualizada: a través de la mutación al nivel del gen, se correlacionan genotipo y fenotipo, para tratar de manera más exacata la mutación que presenten los pacientes.
La fenilalanina es un aminoácido esencial; la de origen dietético, que no se utiliza para sintetizar proteínas, se degrada normalmente siguiendo la vía de la tirosina. El déficit de la enzima fenilalanina hidroxilasa, o de su cofactor, la tetrahidrobiopterina, conduce a la acumulación de fenilalanina en los líquidos corporales. Estas sustancias son tóxicas para el sistema nervioso central y ocasionan daño cerebral.
Durante sus primeros meses de vida, los niños nacidos con FCU aparentan estar sanos; pero si no reciben tratamiento alguno, entre los tres y seis meses comienzan a perder interés por el entorno, y al llegar a la edad de un año, es evidente que padecen un retraso en el desarrollo.
La FCU se hereda cuando ambos padres tienen el gen de la FCU y lo transmiten a su bebé. Cuando uno de los padres tiene el gen de la FCU, pero no padece la enfermedad, se dice que es “portador”. Un portador tiene un gen normal y un gen con FCU en cada célula; su salud no sufre efecto alguno por la presencia de este gen. Cuando ambos padres son portadores, la probabilidad de que ambos transfieran el gen de la FCU a su bebé, y de que este nazca con la enfermedad, es de una entre cuatro (25 %). Dos de cada cuatro bebés heredan el gen de la FCU de uno de sus padres y el gen normal del otro, lo que los convierte en portadores, al igual que sus padres. También hay una probabilidad de una entre cuatro, de que cada uno de ellos le transfiera un gen normal y de que el niño no tenga la enfermedad ni sea portador. Estas probabilidades son iguales durante todos los embarazos.
La fenilcetonuria es una enfermedad que se puede tratar y detectar fácilmente a través de un examen de sangre simple. La mayoría de los estados exigen un examen de tamizaje para todos los recién nacidos, que generalmente se hace con una punción en el talón poco tiempo después del nacimiento.
Debido a que la fenilalanina está comprometida de forma indirecta en la producción de la melanina, el pigmento responsable del color de la piel y del cabello, los niños con fenilcetonuria usualmente tienen un cutis más claro que el de los hermanos no afectados. Estos niños también pueden tener un olor similar al del ratón, que resulta de la acumulación de ácido fenilacético y se puede sentir en el aliento, en la piel y en la orina si la condición no ha sido tratada de inmediato desde el nacimiento, o cuando consumen alimentos que contienen fenilalanina.
Últimamente se han experimentado algunos tratamientos químicos a base de aminoácidos, basados en el efecto ya descrito de que la fenilalanina impide que las neuronas asimilen otros aminoácidos. Se ensayó con complejos de leucina, isoleucina y valina, también con tirosina y triptófano. El resultado fue que su efectividad parece ser útil solo para relajar un poco la dieta durante un corto período de tiempo.
El futuro del tratamiento se proyecta hacia la posibilidad, de que el organismo del niño fabrique una enzima funcionante, lo que ya avanza, gracias a los adelantos en la replicación del ADN. Ya se han obtenido