INTRODUCCIÓN
Costello describe por primera vez en 1977 dos pacientes con características similares: alto peso al nacer, dificultad para alimentarse, facies tosca, pelo ensortijado, papilomas perinasales, piel anormalmente elástica e hiperpigmentada, retraso mental y personalidad humorística.1
Inicialmente se interpretó como un desorden genético raro y complejo, con expresión fenotípica pseudotesaurismótica, asociada fundamentalmente a retraso en el desarrollo físico e intelectual.2
Las referencias actuales lo describen dentro de los síndromes neuro-cardio-facio-cutáneos (SNCFCs), caracterizados por presentar rasgos dismórficos muy semejantes, cariotipo normal y base etiopatogénica e incluso molecular común, determinada por hiperactivación de la vía de transducción Ras-MAPK, secundaria a una mutación en el protooncogén HRAS (83-100 % de los casos).3
Además del síndrome de Costello (SC) dentro de los SNCFCS se incluyen el LEOPARD, el cardio-facio-cutáneo, Noonan, Legius y la neurofibromatosis.3
El síndrome de Costello (SC) es una entidad poco conocida, se reportan aproximadamente 250 casos a escala mundial. Resulta de interés identificar sus manifestaciones clínicas para establecer un diagnóstico precoz, lo que hace posible la estimulación e intervención temprana, así como ofrecer asesoramiento genético a los padres.3,4
Lo anterior fundamenta la pertinencia de presentar un caso de SC, atendido en la provincia de Holguín.
PRESENTACIÓN DEL CASO
Este caso se presenta previo consentimiento informado de los padres para su estudio y publicación. Se trata de un niño de siete años de edad, mestizo y de procedencia rural.
Historia obstétrica anterior: cuatro embarazos (E4), dos partos (P2) y dos abortos (A2) provocados. Alfafetoproteína elevada.
Antecedentes perinatales y postnatales: parto eutócico a término, peso al nacer 2892 gramos y Apgar 8/9. Se ingresó en el servicio de Neonatología por presentar síndrome de dificultad respiratoria secundario a bronconeumonía connatal. Durante su estadía hospitalaria se realizó el diagnóstico clínico y ecocardiográfico de Tetralogía de Fallot, para lo cual recibió posteriormente tratamiento quirúrgico.
Durante los primeros años de vida, presentó dificultad para alimentarse, fallo de medro y retardo en el desarrollo psicomotor. Posteriormente se hizo evidente el retraso mental ligero y un patrón de conducta caracterizado por ser alegre, sociable y apacible.
No se recogieron antecedentes patológicos familiares de interés.
Examen físico
Mensuraciones actuales: peso 15kg, talla 86 cm y circunferencia cefálica (CC) 54 cm.
Evaluación antropométrica (según tablas cubanas de percentiles): peso/edad <3pp, talla/edad <3pp, peso/talla 97pp y CC/edad >97p.
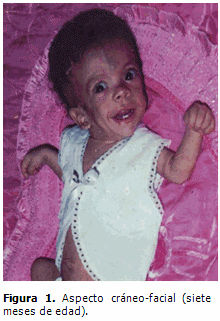
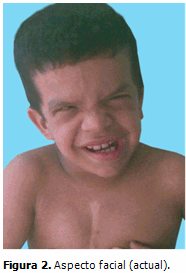
Aspecto cráneo-facial: macrocefalia, facies grotesca, hipertelorismo, orejas de implantación baja con lóbulos prominentes e hipoplásicos, labios gruesos, fosas nasales anchas y pliegue nucal. (Figuras 1 y 2)
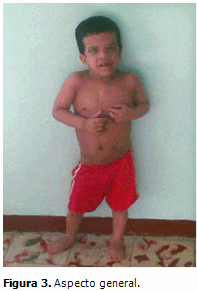
Sistema osteomioarticular: talla baja para su edad y sexo, cicatriz quirúrgica localizada en la línea media de la cara anterior del tórax, hiperlaxitud de las articulaciones interfalángicas y metacarpofalángicas con desviación ulnar de las manos. (Figura 3)
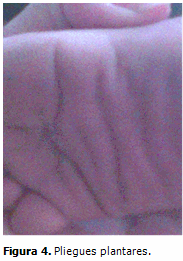
Piel: laxa con pliegues palmo-plantares profundos, coloración oscura a nivel del cuello e hiperqueratósica en manos y pies. (Figura 4)
Aparato cardiovascular: soplo sistólico grado III/VI en borde esternal izquierdo.
Estudios realizados
Cariotipo: 46 XY (varón cromosómicamente normal)
Pruebas metabólicas en orina: negativas. Estas fueron realizadas para descartar errores innatos del metabolismo.
Aminoácidos en sangre normales.
Tomografía axial computarizada (TAC) simple de cráneo: dilatación del sistema ventricular, alteraciones compatibles con el diagnóstico de hidrocefalia.
DISCUSIÓN
El paciente que presentamos tiene peso y talla inferior al tercer percentil para su edad y sexo, debido al déficit de crecimiento ponderal que sufrió en etapas tempranas de la vida, fenómeno descrito en la fase pseudo-marasmática del SC. Actualmente se encuentra en el 97pp del peso para la talla, lo que traduce una recuperación posterior en la ganancia de peso, sin embargo, ya el fallo de medro era irreversible (fase pseudotesaurismótica). Durante este periodo se hicieron más evidentes las características fenotípicas y conductuales que identifican el síndrome, tal y como se refiere la bibliografía consultada.5,6
El aspecto facial se fue tornando cada vez más grosero; el índice de circunferencia cefálica para la edad se mantuvo por encima del 97pp (macrocráneo moderado secundario a hidrocefalia demostrada en TAC), la frente ancha, el pelo ensortijado y frágil, la facies pletórica, epicantos, labios y lóbulos de las orejas gruesos e hipoplásicos y las fosas nasales anchas, caracterizan actualmente sus principales rasgos dismórficos, coincidiendo con lo descrito por la mayoría de los autores.
El retardo en el desarrollo psicomotor, retraso mental ligero y la adquisición de una personalidad humorística, se tornaron indudables a medida que el niño fue ganando en edad. Según Pozo Román, estas constituyen las principales manifestaciones neurológicas del SC.3
La tetralogía de Fallot, la estenosis pulmonar, la comunicación interatrial y la interventricular, son malformaciones del aparato cardiovascular que forman parte de este complejo síndrome, en aproximadamente el 63 % de los casos.1,5,7
La piel de las palmas de las manos y plantas de los pies del paciente en estudio, es laxa, redundante e hiperqueratósica, con pliegues gruesos y acolchonados (aspecto cerebriforme), además de una evidente tendencia a la formación de pliegue nucal. Es innegable la hiperlaxitud de las articulaciones interfalángicas y metacarpofalángicas con desviación ulnar de las manos, como lo describen la mayoría de los autores.2,5,7
En resuman, el paciente exhibe la mayoría de las manifestaciones clínicas que caracterizan el SC, sin embargo, para realizar el diagnóstico clínico fue necesario descartar el resto de las entidades que forman parte de los síndromes neuro-cardio-facio-cutáneos.
Uno de los principales diagnósticos diferenciales, según refiere la mayoría de los autores, es el Síndrome de Nooman (SN), caracterizado por talla baja, estirón puberal reducido o ausente, retardo en el desarrollo psicomotor y retraso mental ligero. Facies triangular con frente ancha, orejas de implantación baja y rotación hacia atrás, hélix grueso, ptosis palpebral, hendiduras palpebrales antimongoloides, proptosis ocular, miopía, nistagmos, micrognatia, cuello corto y palmeado con implantación baja de la línea posterior.3
Descartamos también el Síndrome de LEOPARD, cuyo nombre es un acrónimo que recoge las principales características de la entidad: lentiginosis (máculas planas, de color marrón-negruzco, semejantes a “pecas”, que aparecen alrededor de los 4-5 años y se localizan sobre todo en cara, cuello y parte superior del tronco, incrementándose en número hasta la pubertad), anomalías de conducción en el electrocardiograma, hipertelorismo ocular, estenosis pulmonar, genitales anormales, retraso de crecimiento y sordera neurosensorial.2,3,5
El Síndrome cardio-facio-cutáneo (SCFC) es un raro síndrome de presentación esporádica que, como su nombre indica, se caracteriza por anomalías cardíacas, apariencia facial característica y anomalías cutáneas. Los rasgos fenotípicos cráneo-faciales son muy similares a los del SN, pero con algunos distintivos: macrocefalia, frente amplia y prominente con estrechamiento bitemporal, hipertelorismo, epicantos, fisuras palpebrales antimongoloides, raíz nasal deprimida con narinas ligeramente antevertidas y la punta de la nariz bulbosa. Se podría decir que es una facies semejante a la del Noonan pero más tosca, sin llegar a serlo tanto como la del SC.3 El cuello suele ser corto, a veces con Pterigium colli e implantación baja de la línea posterior del cabello. Son frecuentes las anomalías esternales (Pectus carinatum y/o excavatum). La afectación cutánea incluye: xerosis, hiperqueratosis en brazos, piernas, cara y zonas palmo-plantares, ictiosis, queratosis pilaris, “manchas café con leche”, eccema, nevo pigmentado, lentígines y hemangiomas.3,4 Las alteraciones cardíacas más frecuentes son: estenosis pulmonar, defectos septales y trastornos del ritmo cardíaco. El crecimiento suele estar afectado, generalmente secundario a afecciones gastrointestinales (reflujo gastroesofágico, vómitos, aversión por la comida). Las manifestaciones neurológicas son, prácticamente, constantes: hipotonía, retraso del desarrollo y retraso mental, habitualmente más grave que en SC.5
Síndrome de Legius: previamente conocido como neurofibromatosis tipo 1-like, es un síndrome autosómico dominante, en el que se combinan rasgos de neurofibromatosis tipo 1 (NF1) y el SN. Se caracteriza por: manchas “café con leche”, pecas axilares múltiples y en algunos individuos, un fenotipo facial similar al SN. Las dificultades de aprendizaje y la hiperactividad son frecuentes. A diferencia de la NF1, estos pacientes no tienen nódulos de Lish, neurofibromas, ni tumores del sistema nervioso; mientras que, los lipomas son un hallazgo frecuente.3-5
La tendencia a desarrollar tumores sólidos tanto benignos (papilomas/fibromas en la región perinasal y perianal), como malignos (rabdomiosarcomas, neuroblastomas y carcinomas de vejiga) es una peculiaridad del SC, sin embargo aún no se han manifestado en el paciente en estudio. Generalmente son las últimas alteraciones que aparecen y requieren un seguimiento especial para poder identificarlos tempranamente y adoptar una conducta adecuada.
Se plantea que la activación de la vía de transducción Ras-MAPK, secundaria a la mutación en el protooncogén HRAS, tienen un alto potencial oncogénico; de hecho, la aparición de mutaciones somáticas activadoras en estos genes es muy frecuente y se aplica a más del 20 % de los cánceres descritos en humanos.7-9
El diagnóstico del SC es clínico y se realiza fundamentalmente por las características fenotípicas, las manifestaciones neurológicas y cardiovasculares. Su semejanza con otros síndromes neuro-cardio-facio-cutáneos hace complejo el diagnóstico específico y en la mayoría de los casos hay que esperar que el paciente pase a la fase pseudotesaurismótica, por lo que puede realizarse de forma tardía. El diagnóstico precoz hace posible la estimulación e intervención temprana, la búsqueda activa de lesiones tumorales, así como ofrecer asesoramiento genético a los padres.
