INTRODUCCIÓN
El síndrome hemolítico urémico (SHU) es una afección que se caracteriza por la presencia de anemia hemolítica microangiopática, trombocitopenia e insuficiencia renal aguda.1-5 Es la causa más frecuente de insuficiencia renal aguda en la infancia, la mayoría de los casos (90 %) están asociados a diarrea, siendo frecuente la presencia de colitis hemorrágica. Este grupo, llamado SHU típico, epidémico o diarrea (+), es de buen pronóstico. El 10 % restante corresponde a SHU atípicos, no asociados a diarrea, de inicio insidioso, con tendencia a recaídas y de mal pronóstico. En este grupo se incluyen los secundarios a infecciones no enterales (neumococo, mycoplasma pneumoniae), glomerulopatías primarias, drogas, embarazo, cáncer, SIDA, trasplante de médula ósea y enfermedades del colágeno; también se incluyen las formas idiopáticas y familiares autosómicas dominantes, recesivas y esporádicas.6
El SHU es una complicación poco frecuente de la enfermedad neumocócica invasiva y tiene una mortalidad del 25 %. La mitad de los casos que sobreviven evolucionan ha una enfermedad renal terminal.7,8 Sin embargo, tuvimos la oportunidad de atender a un niño de 17 meses con la enfermedad secundaria a una neumonía con empiema, que después de 10 días en anuria logró una función renal normal. Esto, unido a que no encontramos casos similares publicados en la provincia, motivó la realización del estudio.
PRESENTACIÓN DEL CASO
Paciente masculino de 17 meses, blanco, eutrófico, con antecedentes de embarazo normal y parto a las 42 semanas por cesárea. Peso al nacer de 4350 gramos (macrofeto) y Apgar adecuado. No tuvo problemas en el periparto y presentó una evolución satisfactoria hasta el momento de su ingreso en el Hospital Pediátrico Universitario de Cienfuegos, por fiebre, tos y polipnea.
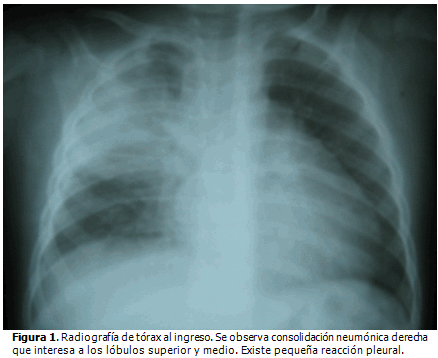
Mediante examen radiográfico se llegó al diagnóstico de neumonía de lóbulos superior y medio derechos (Figura 1). Fue internado en la Unidad de Cuidados Intensivos Pediátricos (UCIP) por encontrarse decaído y con quejido expiratorio importante. Se indicó tratamiento con cefotaxima como antimicrobiano.
Se le realizaron los siguientes análisis de laboratorio:
Hemograma (Hb: 98 g/l, leucocitos: 11,04 X 10 9 /l, stab: 0, segmentados: 66 %, eosinófilos: 0, monocitos: 2 %, linfocitos: 32 %, plaquetas: 160 x109/l).
Hemogasometría (ph: 7,38, PCO2: 32,2 mmHg, PO2: 105 mmHg, HCO3: 20 mEq/l, EB: -5,4 mEq/l, Sat O2: 100 %).
Ionograma (Na: 137mEq/l, CL: 103 mEq/l, Ca: 1,2 mEq/l, K: 4,2 mEq/l), glucemia (5,6mmol/l), creatinina (54 mcmol/l) y urea (2.3 mmol/l).
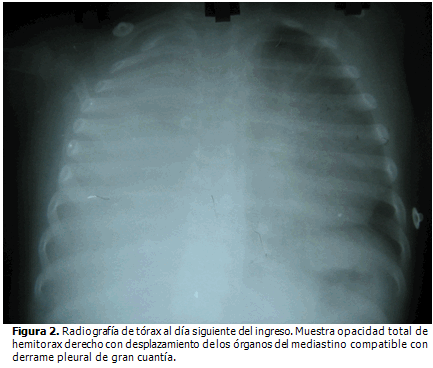
Al segundo día de estancia en la UCIP se observó en una nueva radiografía, empeoramiento de las lesiones, y la presencia de derrame pleural de gran cuantía (Figura 2), por lo que se procedió a realizar pleurocentésis y seguidamente pleurotomía con aspiración continúa, al comprobarse el aspecto purulento del líquido obtenido inicialmente. Además, se añadió vancomicina al tratamiento. Al tercer día de la enfermedad, el estado del paciente se hizo crítico en pocas horas, con sangramiento por los sitios de punción, oliguria, y deterioro de la función respiratoria, siendo necesario intubar y comenzar con ventilación mecánica, así como con apoyo inotrópico con dobutamina. Se comprobó en esos momentos caída de la hemoglobina a 62 g/l y las plaquetas a 12 x109/l, con un tiempo de sangramiento prolongado, por lo que se decidió transfundir glóbulos y plaquetas con urgencia (el resto del coagulograma fue normal). La prueba de Coombs fue negativa. También se comenzó tratamiento con inmunoglobulina G IV en dosis inmunosupresora. Aparejadamente se constató un compromiso renal severo, al recibirse creatinina, urea y acido úrico en cifras de 305 mcmol/l, 27mmol/l y 427mmol/l, respectivamente. Un examen de lámina periférica realizado al día siguiente, mostró presencia de plaquetas desagregadas y escasas, así como crenocitos. Con estos elementos se hizo el diagnóstico de síndrome hemolítico urémico secundario a enfermedad neumocócica. Fue necesario transfundir en varias ocasiones con plaquetas, plasma y hematíes, estos últimos se administraron lavados, excepto la primera vez.
Al cuarto día de evolución, ante la situación renal crítica dada por anuria sin respuesta a diurético, hipertensión arterial, hiperazoemia, acidosis metabólica e hiperpotasemia, se decidió comenzar con diálisis peritoneal en forma de baños continuos, con lo que se logró mejorar inmediatamente el medio interno y se hizo más factible el manejo de los líquidos intravenosos. Para la hipertensión se usó captopril.
Al sexto día de tratamiento, se pudo suspender la ventilación mecánica, y retirar la sonda pleural debido a la mejoría evidente del cuadro radiológico pulmonar. Al aislarse en hemocultivo y líquido pleural, se constató como germen causal al Streptococcus pneumoniae. No se pudo conocer el serotipo, ya que esta prueba no se realiza en nuestro centro.
Fue necesario mantener la diálisis peritoneal continua durante 10 días, en los cuales el paciente estuvo en anuria total; posteriormente apareció diuresis con ritmo progresivo y luego de comprobar un aclaramiento de creatinina aceptable, se suspendió la diálisis. También ascendió la hemoglobina (98g/l) y se normalizaron las plaquetas (180 X109/l).
A los 17 días de estancia en la UCIP, fue trasladado a sala de nefrología previa constancia de una función renal aceptable (creatinina: 141,9 mcmol/l, urea: 10,2 mmol/l, acido úrico: 297,2mml/l, proteinuria de 24 horas: 0,12g/l, aclaramiento de creatinina: 70 ml/mint./1,73 m2). Una semana después fue dado de alta hospitalaria con función renal normal.
DISCUSIÓN
La triada de anemia hemolítica, trombocitopenia y fallo renal agudo permitió hacer el diagnóstico de SHU en el caso presentado. No pudo demostrarse la presencia de esquistocitos en la lámina periférica, lo cual es característico de este síndrome, debido a que fue necesaria una transfusión urgente, sin embargo, se observaron crenocitos. En el SHU se produce un trastorno de la microvasculatura, clínicamente definido por anemia hemolítica microangiopática (negativa en el test de Coombs) y trombocitopenia, que afecta preferentemente a los riñones, y que se manifiesta con hematuria, oligoanuria y fracaso renal. El daño endotelial en la microvasculatura glomerular parece ser el primer episodio en la patogenia del SHU. Este daño se pone de manifiesto por el engrosamiento de la pared vascular, la inflamación del endotelio y su desprendimiento de la membrana basal glomerular. El daño endotelial desata una cascada de acontecimientos que dan como resultado la formación de microtrombos de plaquetas y fibrina, que ocluyen las arteriolas y los capilares renales. La generación de esquistocitos (fragmentos celulares) a causa de la rotura de los eritrocitos que atraviesan esta microvasculatura parcialmente ocluida, es característica del SHU.9
Tradicionalmente, pueden distinguirse dos formas de SHU. La forma más frecuente (en el 90 % de los casos) se denomina SHU clásico o típico y se asocia con diarrea provocada por infección por Escherichia coli, productor de la toxina Shiga (STEC), capaz de unirse a receptores Gb3 (globotriaosilceramida) de la superficie de las células endoteliales y provocar la destrucción de estas de forma directa, o a través de la activación de mecanismos inflamatorios y procoagulantes.10
La mayoría de los pacientes con SHU típico evolucionan satisfactoriamente, solo un 10 % evoluciona hacia una enfermedad renal crónica y un 25 % desarrollan secuelas renales permanentes. El 10 % de los casos restantes presentan el SHU no asociado con diarrea y de peor pronóstico. La mayoría de los pacientes presenta recurrencias y más de un 50 % desarrolla una insuficiencia renal terminal.
Esta forma atípica de SHU tiene una incidencia de aproximadamente 2 casos por millón de habitantes, y una prevalencia de 1/105 niños en la Unión Europea.6 Se produce en ocasiones secundariamente a enfermedad neumocócica, sobre todo neumonía. La fisiopatología se ha vinculado tradicionalmente a la producción de neuroaminidasa, que al unirse al ácido N-acetilneuramínico de las membranas de plaquetas, eritrocitos y capilares glomerulares, provoca la exposición del antígeno Thomsen Freidenreich (antígenoT). La unión de este anticuerpo IgM circulante en plasma, produce aglutinación y hemólisis de hematíes, agregación capilar y alteración de los capilares glomerulares.6,11
Numerosos estudios llevados a cabo en los últimos años, han establecido que el SHU atípico (SHUa) posee un claro componente genético, y que se asocia con frecuencia a mutaciones y polimorfismos en genes que codifican proteínas del sistema del complemento. Se ha demostrado la existencia de una estrecha asociación entre el SHUa y las mutaciones en las proteínas del complemento factor H, MCP, factor I, factor B y C3.12,13
Las mutaciones en factor H asociadas con SHUa son prototípicas del defecto en el complemento que caracteriza a esta enfermedad. El factor H es un regulador que actúa en el plasma, controlando la homeostasis del sistema del complemento, y sobre las superficies celulares evitando el daño a los componentes propios. Las mutaciones en factor H traen como consecuencia que disminuya la protección de las superficies celulares al daño accidental producido por la activación del complemento, aunque no afectan a la regulación del complemento en el plasma.14 El caso presentado no tenía antecedentes familiares de enfermedad renal, por lo que no se asocia este componente genético en la etiología de la enfermedad.
En cuanto al tratamiento, se recomienda realizar de inicio un manejo adecuado de los líquidos y electrolitos, incluyendo la corrección del déficit de volumen, el control de la hipertensión y la temprana instauración de diálisis peritoneal en caso de anuria u oliguria significativa. Según el Grupo de Estudio Pediátrico Europeo, para el SHU se debe iniciar tratamiento con plasmaféresis tan pronto como sea posible, dentro de las 24 horas de presentación en paralelo con el tratamiento anterior, lo que reemplazaría las proteínas defectuosas por proteínas funcionales.15 Además, debido a que existe un desorden de sobreactivación del complemento, una opción terapéutica prometedora, tanto en la fase aguda como en la prevención de las recurrencias, es el uso de anticuerpos monoclonales humanizados, como el C5 (Eculizumab), dirigidos contra los componentes activadores clave de la ruta final del complemento.16,17 Pese a que aun existe debate con respecto a su eficacia debido a que no hay estudios prospectivos controlados randomizados, todas estas medidas han disminuido la tasa de mortalidad de 50 a 25 %.1,18
La transfusión de hemoderivados debe realizarse con hematíes lavados, por la presencia de anticuerpos frente al antígeno T plasmático que agravarían el cuadro.19 En nuestro caso la primera transfusión fue de hematíes no lavados debido a la urgencia de la situación. Tampoco pudimos realizar plasmaféresis porque no disponemos del equipamiento ni de los recursos que se requieren para ello. En cuanto al uso de inmunoglobulina G IV, hay publicaciones a favor de su uso, si bien no existen estudios que demuestren de manera consistente su utilidad.20 Desconocemos si en la evolución favorable del paciente influyó el empleo de esta terapéutica.
Puede concluirse que el éxito en el tratamiento del caso que presentamos estuvo dado por la rápida identificación de la enfermedad, la monitorización de las complicaciones y la meticulosidad en el apoyo vital, incluyendo la realización de diálisis precoz intensiva.
