INTRODUCCIÓN
La anemia falciforme es una enfermedad genética de herencia autosómica recesiva, asociada con episodios de dolor agudo y daños progresivos en diferentes órganos. (1) Fue la primera enfermedad de origen molecular reportada en el mundo, y en la actualidad es la hemoglobinopatía más frecuente, con más de 250 millones de portadores a nivel mundial. (2)
La anemia drepanocítica, como también se le conoce, es causada por mutaciones puntuales en el gen HBB, localizado en el brazo corto del cromosoma 11, locus 15.5. La variante alélica βS, originada por una sustitución de adenina por timina en el sexto codón del gen de β globina, es la causa más común de anemia falciforme, (3) resultando la más frecuente en los africanos y su descendencia. (2) El resultado de esa sustitución provoca la aparición de un residuo de valina en lugar de ácido glutámico, por lo que al ser este un aminoácido apolar, surge un “parche” hidrofóbico en la superficie de la molécula. Esta alteración tiene poco efecto sobre la solubilidad de la hemoglobina S oxigenada, pero sí se reduce significativamente en la molécula desoxigenada. (4) Por otra parte, la variante alélica βC (HbC), es producto de la sustitución de guanina por adenina, lo que genera que en el ARNm se encuentre codificado el aminoácido lisina en lugar de ácido glutámico. (5) Aunque la frecuencia de esta mutación es menor, el número de adultos afectados con la variante alélica βC en doble dosis es casi igual que el de los adultos SS, debido a que los pacientes con genotipo CC tienen una supervivencia cercana a la normal, ya que la anemia hemolítica característica de esta hemoglobinopatía es ligera. (6,7)
En Cuba, la frecuencia de portadores para esta enfermedad oscila entre el 3 % y el 10 % en las diferentes regiones, siendo mayor en algunas provincias orientales como Santiago de Cuba. Desde la década de los 80 se estableció el Programa de prevención de anemia falciforme en el país, que permite la pesquisa de todas las gestantes mediante la realización de electroforesis de hemoglobina a la captación del embarazo, con el objetivo de identificar las parejas con riesgo de tener descendencia afectada por esta hemoglobinopatía y brindarles, como parte del asesoramiento genético, la opción del diagnóstico prenatal de la anemia falciforme. (8)
El método de elección para la búsqueda de las mutaciones βS y βC es PCR-ARMS (del inglés: polymerase chain reaction y amplification refractory mutation system); aunque todos los posibles genotipos SS obtenidos por este métodos son confirmados por la técnica de PCR-RFLP (del inglés: restriction fragment lenght polymorphism), empleando los cebadores PCO1-PCO9.
Debido a que la anemia falciforme constituye un problema de salud a nivel mundial, principalmente en los países con ascendencia africana, entre los cuales se encuentra Cuba, esta investigación tiene el propósito de determinar la frecuencia de las mutaciones βS y βC en los pacientes cubanos estudiados en el año 2010, corroborando mediante la técnica PCO1-PCO9 los potenciales genotipos SS obtenidos por el método de PCR-ARMS.
MÉTODOS
Se realizó un estudio descriptivo, de una muestra conformada por 270 fetos de parejas portadoras de los alelos S y/o C, procedentes de diferentes provincias de Cuba, cuyas muestras fueron enviadas desde los Centros Provinciales de Genética al laboratorio de Biología Molecular del Centro Nacional de Genética Médica, en el año 2010, para el estudio molecular de anemia falciforme, previo asesoramiento genético y consentimiento informado de las gestantes.
Extracción de ADN a partir de líquido amniótico
La extracción de ADN fue realizada por el método de precipitación salina, descrito por Miller SA, en 1986, (9) a partir de una muestra de 20mL de líquido amniótico, recolectado en un tubo estéril.
Amplificación de los alelos βA, βS y βC
Las condiciones del método de PCR-ARMS para la amplificación de los alelos A, S y C fueron las siguientes: 100 ng de ADN, 1,5 µM de los cebadores GHPCR1, GHPCR2 (controles internos de amplificación) y BGP2 (cebador común para amplificar los alelos βA, βS y βC diseñados por Wu y col.(19)), dNTP a 0,1mM, 1X del tampón correspondiente a la polimerasa utilizada y 1,0 mM de MgCl2, para un volumen final de 25 µl, completado con 0,85 µM de uno de los cebadores βA, βS y βC, según el alelo correspondiente a cada tubo de reacción, y 1U de Taq ADN polimerasa por reacción, añadida a la mezcla luego del primer ciclo de desnaturalización. La amplificación se efectuó en un termociclador MJ Research, e incluyó un primer ciclo de desnaturalización de 94°C por 4 minutos, seguido de 29 ciclos de: 1 minutos a 94 oC, 2 minutos a 55 oC y 1 minuto a 72 oC, y una extensión final de 5 minutos a 72oC.
Una vez culminado el proceso, se mezclaron los 25ml del producto de PCR y 5ml de bromofenol azul (BFA) como frente de corrida, y fueron aplicados en un gel de agarosa al 2 % teñido con bromuro de etidio. Se utilizó el tampón TBE a 0,5X (0,5 M de Tris, 10mM de EDTA, 0,5mM de ácido bórico) para la corrida electroforética que se realizó durante 30 minutos a 250 Voltios. Las bandas obtenidas se visualizaron en un transiluminador al exponerlas a luz ultravioleta.
Amplificación del fragmento β-globina por la técnica PCO1-PCO9 y digestión enzimática.
Las condiciones utilizadas en este método fueron las siguientes: 100ng de ADN, 1U de Taq ADN polimerasa por reacción, 1X del tampón de la enzima, 0,75 µM de los cebadores PCO1 y PCO9, dNTP a 0,2 mM, MgCL2 a 1,5 mM en un volumen final de 50µl. Las condiciones del PCR fueron 5 minutos a 94oC, seguido de 30 ciclos de 45 segundos a 94oC, 45 segundos a 55oC, 45 segundos a 72oC y una extensión final de 5 minutos a 72oC en un termociclador MJ Research. La digestión enzimática se efectuó en un volumen final de 25μL, que contenía 10 U de la enzima de restricción Bsu 36.I, 1X del tampón de la enzima, 1X de albúmina de suero bovino (BSA) y 10µL del producto amplificado, incubándose a 37°C por 4 horas. Posteriormente se mezcló con 5μL de BFA y se aplicó en un gel de agarosa al 2 %, siendo sometido a una corrida electroforética con el tampón de corrida TBE 0,5X, a 250 Voltios durante 40 minutos. Finalmente, se visualizaron los fragmentos al exponer el gel de agarosa teñido con bromuro de etidio, a luz ultravioleta.
Análisis estadístico
Para el cálculo de la frecuencia génica se utilizó una formula derivada de la Ley de Hardy-Weinberg, donde F representa la frecuencia alélica, p el alelo objeto de estudio y q cualquier otro alelo presente en la muestra:
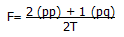
RESULTADOS
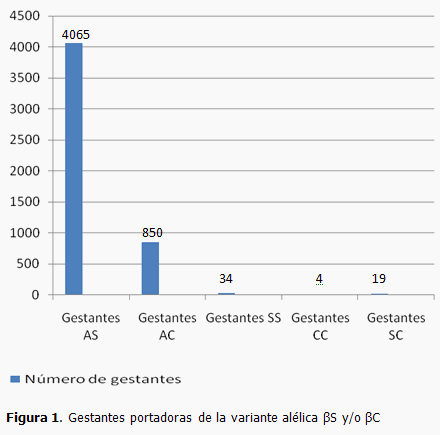
En el año 2010 fueron detectadas 4972 gestantes portadoras de la variante alélica βS y/o la βC en estado heterocigótico, heterocigótico compuesto u homocigótico (Figura 1), mediante el programa nacional de detección de portadoras de anemia falciforme. Además se estudiaron 4622 esposos de esas gestantes, e identificados 312 con riesgo de tener un niño con anemia falciforme. Un total de 270 parejas aceptaron la opción de diagnóstico prenatal molecular, lo que representó un 86,5% de las parejas de riesgo estudiadas.
De los 47 fetos examinados, nueve resultaron ser en realidad heterocigotos para el alelo S (AS), lo que arrojó una exactitud del 80,8 % del método de PCR-ARMS al discriminar los genotipos AS de los SS.
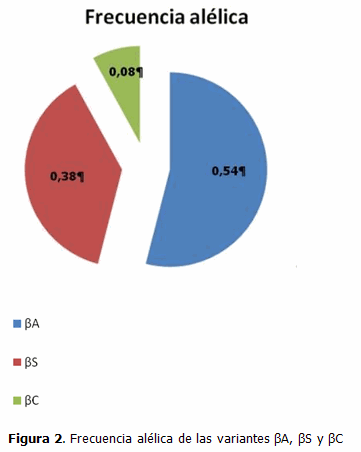
De las muestras estudiadas, 210 presentaron el alelo βA, de los cuales 83 lo mostraron en estado homocigótico y 127 heterocigótico, lo que arrojó una frecuencia alélica de 0,54 para este alelo. Por otra parte, el alelo βS fue hallado en 164 muestras de los fetos estudiados, siendo 38 de ellos homocigóticos para esa mutación, 21 heterocigóticos compuestos y 105 heterocigóticos, con una frecuencia alélica de 0,38 en la población estudiada. El alelo βC se encontró en 44 fetos, de los cuales solo 1 resultó ser homocigótico para esta mutación, 21 heterocigóticos compuestos y 22 heterocigóticos, proporcionando una frecuencia alélica de 0,08 para el alelo βC. (Figura 2).
A las 60 gestantes que se les detectó un feto afectado con anemia falciforme, se les brindó asesoramiento genético; de ellas, 45 optaron por la interrupción de la gestación.
DISCUSIÓN
Los 270 fetos fueron estudiados por el método de PCR-ARMS, mientras que por la técnica conocida por PCO1-PCO9, se amplificó un fragmento del gen β-globina correspondiendo a una talla de 687pb, en los 47 fetos con posible genotipo SS obtenido mediante el método anterior, debido a que en ocasiones no amplifica el alelo A por este procedimiento a pesar de estar presente en su genoma, aparentando un genotipo SS. Esta técnica de PCR-RFLP convencional solo requiere una reacción por muestra, por lo que con respecto al ADN necesario para su realización, aventaja a la técnica anteriormente expuesta, la cual utiliza entre dos y tres veces mayor cantidad de material biológico.
La técnica de PCR-ARMS, ha constituido una ventaja desde el punto de vista social y económico para el sistema nacional de salud y para la población cubana, pues permite un ahorro significativo de divisas al país (1,10 dólares por muestra) al no utilizar métodos que requieren enzimas de restricción, además de favorecer la obtención de resultados con mayor brevedad. En el periodo analizado, ha posibilitado la realización del 82,5 % de los diagnósticos prenatales de anemia falciforme.
Sin embargo, este método presenta como desventaja la existencia, en ocasiones, del fenómeno de enmascaramiento en la amplificación diferencial del alelo A, por lo que resulta necesario comprobar los resultados en los casos con posible genotipo SS con la técnica PCO1-PCO9, en la que se digiere, luego de la amplificación de la región de interés, con la enzima de restricción Bsu36 I, para discriminar entre la presencia del alelo S y los alelos A o C. Por otra parte, concordando con los resultados encontrados en otras poblaciones, la frecuencia alélica del alelo βC en la población cubana es baja, (2) ya que en el periodo analizado solo se diagnosticó un feto homocigótico para este alelo.
El cuadro clínico de estos pacientes se caracteriza por molestias abdominales, artralgias, cefaleas, esplenomegalia en el 90 % de los casos sin afectación de la función esplénica, y algunos casos pueden presentar colelitiasis. El diagnóstico de laboratorio indica la presencia de anemia ligera a moderada, reticulocitos (entre 3 y 6 %) en sangre periférica, dianocitos y cristales de hemoglobina C, mientras que la electroforesis de la Hb señala que la Hb C y la Hb fetal están ligeramente aumentadas con ausencia de Hb A. (11) No obstante, los pacientes pueden llevar una vida relativamente normal.
En la población estudiada, el genotipo SC resultó ser menos frecuente que el genotipo SS, lo que guarda relación con el comportamiento del alelo βC en la población cubana anteriormente descrita. (12) El cuadro clínico de estos pacientes es algo menos severo que el de la hemoglobinopatía SS; presentan esplenomegalia moderada que puede persistir en la vida adulta con alteración de su función e infecciones. Se han descrito tres complicaciones fundamentales: retinopatía proliferativa, necrosis aséptica de la cabeza del fémur y síndrome torácico agudo. (11)
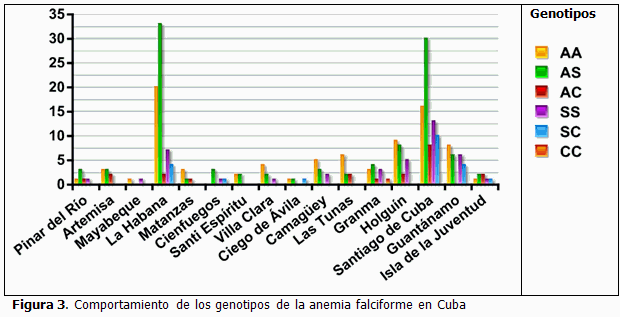
Es relevante el número de portadoras identificadas en las provincias de La Habana y Santiago de Cuba, territorios donde resultó más elevado con respecto al resto del país (Figura 3), lo que coincide con datos que refieren la presencia de una mayor cantidad de portadores de anemia falciforme en estas provincias, (12) con una frecuencia y una frecuencia estimada del alelo βS de 3,04 % y 5,59 %, respectivamente. La alta frecuencia encontrada en estas zonas está dada por la existencia de una gran población de personas de piel negra, surgida a partir de la trata de esclavos existente en la época colonial de la isla, así como a las migraciones mayormente haitianas hacia la ciudad de Santiago de Cuba, mientras que los resultados obtenidos en La Habana se corresponden con la migración de los nativos de Santiago de Cuba hacia la capital de la isla, y con la alta frecuencia de mestizaje que existe, teniendo en cuenta además que estas provincias son de las más pobladas de nuestro país.
Genotipos
Por otra parte, las provincias de Ciego de Ávila, Sancti Spíritus, Matanzas y Cienfuegos reportan, las frecuencias más bajas de portadores de anemia falciforme, lo cual pudiera estar en correspondencia con la composición y el origen ancestral de su población. (14, 15)
Según los resultados obtenidos en este periodo, han ocurrido 210 nacimientos correspondientes a niños portadores de Hb S ó Hb C, los cuales en un futuro, si así lo desean, serán asesorados genéticamente con respecto a su condición de portadores de anemia falciforme. Los 270 diagnósticos prenatales de anemia falciforme realizados en el año 2010, muestran que sin la existencia de este programa nacional de detección de portadoras de anemia falciforme, hubiesen nacido 60 niños con esta enfermedad sin el conocimiento de sus padres sobre la existencia de la afección, ni la adecuada preparación del sistema de salud para brindarles una atención temprana y diferencial.
