INTRODUCCIÓN
El síndrome de Proteo (SP) fue descrito como entidad nosológica por primera vez en 1979 por Cohen y Hayden, sin embargo no recibió la denominación hasta 1983 en un artículo publicado por Wiedemann et al, para describir cuatro niños con malformaciones hamartomatosas congénitas.1
Wiedemann utilizó como referencia, para abordar las alteraciones estructurales que definen el síndrome, a un legendario héroe griego, transformado por los dioses en un demonio inmortal del mar, el cual podía cambiar de forma a voluntad, y que fue descrito en La Odisea por Homero.2
Es considerado un raro desorden genético que se produce por mutación en el gen 10q23.31, encargado de activar de forma esporádica el crecimiento de varios tejidos (epidérmico, conectivo, óseo, adiposo, endotelial) durante el periodo de desarrollo embrionario, por lo que se manifiesta generalmente al nacimiento o en los primeros años de vida y se caracteriza por gigantismo parcial de manos y pies, nevos pigmentados, hemihipertrofia cutánea, tumores hamartomatosos subcutáneos, macrocefalia y anomalías craneales.3-5
Desde su primera descripción hasta la actualidad solo se han diagnosticado poco más de 500 casos en países desarrollados, con afectación por igual de ambos sexos. Constituye una enfermedad progresiva que conlleva a desfiguraciones severas, limitaciones físicas y reducción de la calidad de vida.6-8
No se encontraron referencias en las publicaciones médicas cubanas donde se describa la existencia de pacientes con esta extraordinaria entidad clínica por lo que el objetivo de este trabajo es presentar un caso diagnosticado con el síndrome de Proteo en Moa, provincia Holguín.
PRESENTACIÓN DEL CASO
Escolar de sexo masculino de ocho años de edad, color de piel negra, procedencia urbana, nacido de parto distócico por cesárea, pesó al nacer 2 830 gramos, apgar 9/9, sin antecedentes patológicos prenatales o familiares de interés.
Al nacimiento se presentó hemihipertrofia de la región glútea, pierna y pie izquierdo, con macrodactilia, displasia del desarrollo de cadera, genusvalgo bilateral y hemangioma plano en abdomen y espalda.
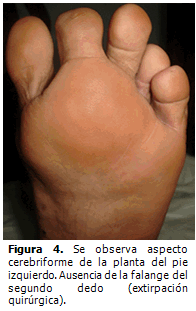
A los dos años de edad se detectó escoliosis dorso-lumbar y cifosis dorsal pronunciada, hiperplasia de la musculatura de la región glútea y muslo izquierdo con aumento de tejido celular subcutáneo y tejido adiposo a nivel dorsal. Tres años después fue necesario realizar extirpación quirúrgica de la falange distal del segundo dedo del pie izquierdo por hipertrofia cortical marcada que le imposibilitaba ponerse zapato.
Todas las malformaciones anteriores se pueden observar en las siguientes figuras. (Figuras 1,2,3).
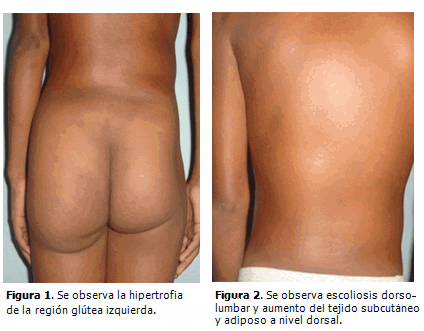
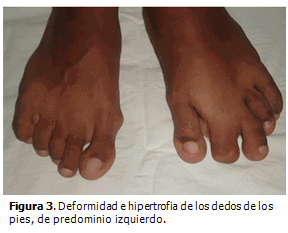
En la actualidad es evidente la progresión de la enfermedad. Presenta deformidad de los huesos del metatarso y de las falanges de los dedos del pie izquierdo así como aspecto cerebriforme de la piel que cubre la planta del pie e hiperostosis de las mastoides. (Figura 4).
Otros datos de interés fueron la aparición de máculas hipercrómicas con bordes bien definidos en el dorso y dilatación venosa en la región hipogástrica de la pared abdominal.
DISCUSIÓN
Se han postulado diversas propuestas de criterios diagnósticos para el síndrome de Proteo, las cuales se basan fundamentalmente en parámetros clínicos. Los primeros fueron propuestos por Wiedemann y colaboradores, en 1983, en una serie inicial de cuatro pacientes,2 aunque ninguno de ellos presentaba la totalidad de la sintomatología descrita.
Costa y colaboradores, en 1985,9 y Clark,10 en 1987, dividen los signos clínicos del SP, según su frecuencia, en principales, frecuentes y ocasionales.
Posteriormente, Samlaska et al,11 en 1989, con una serie de 34 pacientes y Sayama et al,12 en 1994, en una revisión de 63 casos, proponen una relación según los hallazgos clínicos más frecuentes. Para estos autores los criterios frecuentes eran los encontrados en más de la mitad de los pacientes.
Hotamisligil,13 en 1990, fue más estricto dando una puntuación a cada sintomatología. Así, otorga a la macrodactilia y/o hemihipertrofia, cinco puntos; al engrosamiento palmar o plantar, cuatro puntos; los tumores subcutáneos y lipomas, cuatro puntos; los nevos epidérmicos, tres puntos; la macrocefalia o las exostosis craneales 2,5 puntos y al resto de anormalidades menores un punto.
El diagnóstico se establecía si el paciente sumaba 13 o más puntos.
Para clarificar todos estos criterios, en marzo de 1998, se convocó la primera conferencia nacional del SP para padres y familiares (National Institutes of Health de Bethesda, Maryland, EE.UU.), donde se establecieron una serie de criterios diagnósticos, recomendaciones, guías de evaluación y diagnósticos diferenciales.14
Como criterios generales (obligatorios) se incluyeron la distribución de las lesiones en mosaico, el curso progresivo y la aparición esporádica, mientras que los criterios específicos se clasificaron en tres categorías A, B y C. (Tabla 1). El criterio incluido en la categoría A, en presencia de todos los criterios obligatorios es suficiente para el diagnóstico, así como dos de la categoría B o tres de la C.14
El paciente que se presenta en este trabajo reúne los tres criterios obligatorios así como dos de los criterios específicos incluidos en la categoría B (nevus epidérmicos, crecimiento desproporcionado de miembros e hiperostosis mastoidea) y dos de la categoría C (tejido adiposo disrregulado y malformaciones vasculares) por lo que se puede concluir que padece de síndrome de Proteus.

Como diagnósticos diferenciales es necesario destacar que se descartaron otras enfermedades hamartomatosas como la neurofibromatosis, los síndromes de Kipplel-Trenaunay, Mafucci, Bannayan-Riley-Ruvalcaba, Parkes-Weber, del nevo epidérmico, la lipomatosis simétrica y lipomatosis encefalocraneocutánea.1-4
La mayoría de los casos de síndrome de Proteo, se clasificaban como neurofibromatosis de Von Recklinghausen hasta que se establecieron los criterios diagnósticos de los dos tipos de neurofibromatosis en 1988. Sólo dos años antes, los antropólogos Tibles y Cohen habían identificado el síndrome de Proteus como el trastorno que padeció Joseph Merrick, “el hombre elefante”, considerado hasta entonces como un caso de neurofibromatosis.1,7
