INTRODUCCIÓN
La hemocromatosis tipo I (OMIM 235 200) es el defecto congénito del metabolismo más frecuente en las personas con ascendencia europea. Estudios de población basados en ensayos fenotípicos que utilizan la sobrecarga férrica como marcador, muestran que en la población caucásica se encuentra afectado 1 de cada 200 a 400 individuos. (1) La enfermedad tiene un patrón de herencia autosómico recesivo y se caracteriza por una elevada absorción de hierro a nivel de las criptas intestinales como consecuencia de una alteración del metabolismo de este mineral, lo que provoca su depósito progresivo en distintos órganos, como el hígado, páncreas, corazón, articulaciones y otros. (2,3)
La incidencia en Cuba de la hemocromatosis aún no ha sido reportada. A pesar de que recientemente se logró introducir en el país el diagnóstico molecular de esta enfermedad, el diagnóstico se continúa realizando mediante el cuadro clínico del paciente y la cuantificación de hierro y ferritina séricos, de forma que no se consigue identificar a los pacientes asintomáticos ni a los portadores. (2,3)
El gen de la hemocromatosis tipo I se denomina HFE y se localiza en el brazo corto del cromosoma 6, en el locus 6p21,3 donde se han identificando varias mutaciones asociadas a la enfermedad. (2,4) Las principales son la sustitución de una cisteína por una tirosina en posición 282 de la proteína (C282Y) y el cambio de una histidina por un aspártico en posición 63 (H63D). (4-6)
Existe un tratamiento paliativo que puede frenar la evolución de la hemocromatosis tipo I, (6,7) lo que justifica que se intente realizar un diagnóstico precoz antes de que se produzca el daño irreversible (cirrosis, hepatocarcinoma, miocardiopatía, diabetes mellitus, impotencia, artritis), logrando que la esperanza de vida de estos pacientes sea equiparable a la de la población normal, razones por las cuales se realizó esta revisión bibliográfica.
DESARROLLO
Reseña histórica
En 1865, el médico francés Armand Trousseau describió un síndrome clínico caracterizado por cirrosis hepática, diabetes e hiperpigmentación cutánea, en un varón de 28 años; era relevante el tono oscuro de la piel y en especial el color negruzco del pene. A esta triada Trousseau la denominó diabetes bronceada por su asociación con la hiperpigmentación cutánea tan llamativa de los enfermos.(8)
En 1889, von Recklinghausen utilizó el nombre de hemocromatosis para designar el síndrome clínico de Trousseau, tras descubrir que el hierro era el metal acumulado en los tejidos y conjuntamente sospechar el carácter hereditario del proceso al observar la agregación familiar de sus casos. El Dr. Joseph H. Sheldon, afianzó los estudios de Recklinghausen al revisar los trabajos sobre la agregación familiar de la hemocromatosis en 14 autores y confirmó en 1935 que se trataba de una enfermedad genética que altera el metabolismo del hierro. (9)
La naturaleza genética de la enfermedad se demostró 40 años más tarde por Simon y colaboradores en 1975, cuando publicaron su investigación sobre la asociación con el alelo HLA-A3 del complejo mayor de histocompatibilidad en el cromosoma 6. (10) Posteriormente, en 1996 John N. Feder y colaboradores identificaron el gen responsable y las dos mutaciones principales asociadas a esta entidad, lo que marcó un hito en el entendimiento y manejo de la enfermedad, así como del metabolismo del hierro, (11) John nombró al gen HLA-H, aunque más tarde pasaría a denominarse HFE.
Barton y colaboradores identificaron en 1999 cuatro nuevas mutaciones del gen HFE en pacientes clínicamente diagnosticados con hemocromatosis tipo I. En la actualidad se han descubierto más de 20 mutaciones asociadas a esta enfermedad. (2,5,11)
Epidemiología
La hemocromatosis tipo I es la enfermedad genética más frecuente en los individuos de color de piel blanca, afecta a 1 de cada 200 personas. (2,4,5,12) Todos los estudios han demostrado su mayor prevalencia en individuos de raza blanca, de origen caucásico y céltico. La migración de los pueblos celtas explicaría, quizás, la diseminación de la enfermedad por todo el mundo. (1,13)
Además de las diferencias étnicas, el género también parece guardar relación con la enfermedad. Los hombres la padecen con mayor frecuencia que las mujeres, en una proporción 3:1. Factores fisiológicos femeninos, como son la menstruación o el embarazo, impiden la acumulación de grandes cantidades de hierro en el organismo, lo que es una de las causas de dicha discordancia. (14)
Se transmite de forma autosómica recesiva, esto significa que en una pareja de portadores, su descendencia tendrá una probabilidad de un 25 % de hijos sanos, 50 % de hijos portadores y 25 % de hijos con ambos alelos del gen HFE mutados. (2,4,5) La incidencia de heterocigocidad para el alelo de la hemocromatosis en la población blanca es de aproximadamente el 10 %. La incidencia esperada de homocigocidad es de 2-3 por 1000 individuos. (2,4,5) Su gran prevalencia sugiere que ha existido una selección positiva, para mutaciones del gen HFE o para el haplotipo ancestral del complejo mayor de histocompatibilidad conservado con el que se asocia la mutación. Es una enfermedad que resulta en un exceso de hierro, por lo que pudiera conferir una ventaja selectiva en periodos de déficit nutricional de hierro o en estadios fisiológicos que requieren más hierro, como en el embarazo. (15)
Hemocromatosis hereditaria tipo II o hemocromatosis hereditaria juvenil
Es una forma rara y grave de hemocromatosis hereditaria (16) (OMIM 602 390) que afecta a niños o adultos jóvenes de menos de 30 años de uno y otro sexo. Se muestra en sujetos de raza caucásica y procedencia europea y se define clínicamente por presentar hipogonadismo hipogonadotropo, miocardiopatía, hepatomegalia, cirrosis hepática y pigmentación melánica de la piel. Presenta una herencia autosómica recesiva y en la mayoría de los casos está ligada al cromosoma 1q, en que se denomina hemocromatosis tipo IIa; en una minoría de casos la enfermedad está ligada al cromosoma 19, entonces se conoce como hemocromatosis tipo IIb.
Hemocromatosis hereditaria del tipo III asociada al receptor 2 de la transferrina
La hemocromatosis tipo III (17) (OMIM 604 250) está asociada a mutaciones en el gen del receptor 2 de la transferrina (TfR2). Es frecuente que los pacientes presenten sintomatología a edades tempranas, incluso antes de los 30 años, que existan manifestaciones cardiacas, hipogonadismo hipogonadotrópico, artralgias, hiperpigmentación de la piel y cirrosis hepática. El cuadro puede recordar al de la hemocromatosis tipo II.
Hemocromatosis hereditaria tipo IV o enfermedad de la ferroportina
La hemocromatosis tipo IV (18) (OMIM 606 069) se transmite de forma autosómica dominante y es común que la tasa de ferritina en sangre esté muy elevada, sin que este aumento se acompañe de una saturación paralela de la transferrina. En estos casos no se observa el predominio periportal de la siderosis hepatocitaria habitual en la hemocromatosis clásica. Esta siderosis es bien tolerada y la fibrosis hepática es leve o inexistente. Mutaciones en el gen de la ferroportina-1 (FP-1) situado en el cromosoma 2q32 son responsables de esta enfermedad.
Manifestaciones clínicas
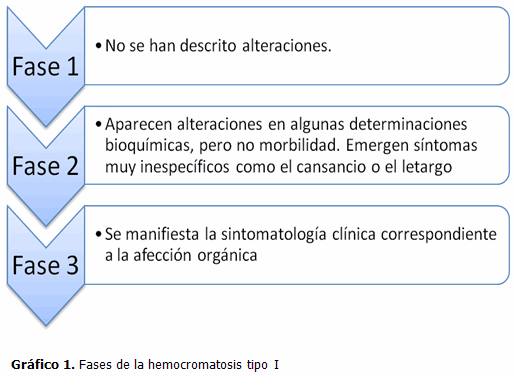
Los individuos con un genotipo asociado a hemocromatosis tipo I no siempre expresan la enfermedad. En los casos con manifestaciones clínicas, la expresión patológica del fenotipo ocurre de acuerdo a las siguientes fases: (6, 19,20)
Fase 1: del nacimiento a la adolescencia. No se han descrito alteraciones.
Fase 2: de la adolescencia a la quinta década de la vida. Aparecen alteraciones en algunas determinaciones bioquímicas que posibilitan el diagnóstico bioquímico. Todavía no se presentan lesiones orgánicas y, por tanto, el sujeto no puede aún considerarse “enfermo”.
Fase 3: a partir de la quinta década de la vida. Se inicia un proceso de daño parenquimatoso y aparecen las “enfermedades asociadas”: hepatopatía, cirrosis hepática, carcinoma hepático, diabetes mellitus, insuficiencia cardíaca, impotencia, artropatía, oscurecimiento cutáneo.
Sólo los individuos que llegan a la fase 3 pueden considerarse enfermos. El resto tienen marcadores, primero los genéticos y después los bioquímicos, que son predictivos de una presentación futura de enfermedad si ello no se evita. (Gráfico 1).
La hemocromatosis debe diagnosticarse en fases pre-clínicas, (1 y 2) mediante análisis genéticos o bioquímicos. Algunos pacientes se diagnostican tardíamente, a partir del conjunto de síntomas de la enfermedad, lo que supone un fracaso. El paciente con hemocromatosis tipo I diagnosticado en fase asintomática podrá evitar todas las complicaciones orgánicas graves de la enfermedad mediante un tratamiento sencillo, barato y seguro que es la flebotomía. (2,4,5,21)
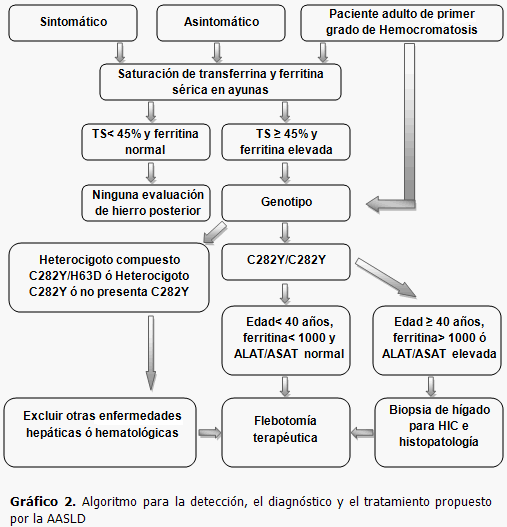
El diagnóstico de la hemocromatosis requiere:
1) Sospecha por análisis sugestivo en paciente con o sin clínica (presencia de uno de los siguientes datos analíticos o varios en dos ocasiones separadas un mínimo de 3 meses): (7)
- Hierro sérico ≥ 150 μg/dL
- Índice de saturación de transferrina > 45 %
- Ferritina sérica > 400 μg/L
2) Demostración de la presencia de las mutaciones más frecuentes del gen HFE.
3) Posteriormente, es conveniente la confirmación de la acumulación de hierro mediante resonancia magnética nuclear o biopsia hepática en caso de que la ferritina sérica sea mayor de 1000μg/L.
Las pautas de la Asociación Americana para el Estudio de las Enfermedades Hepáticas (AASLD por sus siglas en inglés) para el diagnóstico y tratamiento de hemocromatosis hereditaria establecen que “se debe exhortar en forma contundente a todos los pacientes con hemocromatosis que tengan evidencia de sobrecarga de hierro a someterse a flebotomías regulares hasta que los depósitos de hierro se reduzcan”. (Gráfico 2).
Etiología
La causa molecular que provoca la hemocromatosis tipo I son las mutaciones en el gen HFE que codifica la proteína del mismo nombre. (2,4,5)
Genética
El gen HFE se localiza en el brazo corto del cromosoma 6 en el locus 21.3, organizado en 7 exones. Su ARNm, de cerca de 2700 pb de longitud, codifica la proteína HFE. (2,4,5)
Proteína HFE
El gen HFE codifica para una glicoproteína de 343 aminoácidos, muy similar en su estructura tridimensional a la de las moléculas del complejo mayor de histocompatibilidad de clase I. Esta proteína tiene seis dominios diferentes: una cadena peptídica de 22 residuos, tres dominios extracelulares alfa en forma de bucle, una región transmembranosa y un corto residuo C-terminal intracelular que le sirve a la proteína de anclaje en la membrana celular. La estructura terciaria de esta proteína se mantiene gracias a puentes disulfuro. Entre los bucles alfa 1 y alfa 2 queda un espacio por el que interactúa con los receptores de la transferrina. El bucle alfa 3 formado mediante un puente tiol entre dos moléculas de cisteína es fundamental para la unión no covalente a la beta 2 microglobulina y para la expresión de la proteína en la superficie de las células. (2)
Esta proteína se encuentra físicamente donde hay receptores de la transferrina tipo 1, como por ejemplo las células epiteliales de las criptas duodenales y el sincitiotrofoblasto. La proteína puede localizarse en todo el tracto digestivo pero su máxima densidad se ha observado en las criptas de los enterocitos duodenales. También se encuentra su expresión en las células epiteliales del tracto biliar, en las células de Kuppfer hepáticas, en las células del sincitiotrofoblasto, en macrófagos, granulocitos circulantes y monocitos.
Función de la proteína HFE
La proteína HFE se asocia, en dependencia del pH, con el receptor de la transferrina, disminuyendo la afinidad de este por la transferrina cargada, con la que compite por su unión al receptor. Debido a su vinculación con la vía de incorporación de hierro mediada por la transferrina y su localización en los endosomas y la cara basolateral de los precursores enterocíticos, se plantea que la HFE puede ser un sensor de las reservas corporales de hierro, e incluso se ha sugerido que la relación de la HFE con el receptor de la transferrina es crítico para el mantenimiento de la homeostasia del hierro. En fecha reciente, se ha sugerido que la HFE normalmente facilita más que obstaculiza la incorporación celular de hierro unido con la transferrina mediada por el receptor de la transferrina, y parece que la HFE puede también unir otras proteínas o ejercer un efecto directo sobre el transporte endosomal del mineral. (22)
Mecanismo fisiopatológico
La cantidad de hierro total del organismo es de unos 4-5 g en individuos sanos y se mantiene dentro de esos límites durante toda la vida, gracias a que su absorción intestinal se encuentra sometida a un estrecho control. En los pacientes con hemocromatosis este equilibrio está roto por lo que la absorción intestinal de hierro se ve muy aumentada. (Tabla 1).

Existen dos mecanismos que explican cómo tiene lugar este desorden homeostático: mediante el papel de las criptas intestinales y el de la hepcidina. (19,20)
La hipótesis más aceptada es la relacionada con el papel de la hepcidina, a partir del mayor conocimiento de su funcionamiento y su asociación con la hemocromatosis juvenil. Cuando la sideremia es elevada, la síntesis de hepcidina aumenta y disminuye la liberación de hierro de los enterocitos y de los macrófagos, posiblemente gracias a la interacción de otras proteínas relacionadas con la homeostasis del hierro y que aún no están completamente estudiadas, entre ellas la ferroportina. Cuando la sideremia desciende lo hacen también los niveles de hepcidina lo que provoca un aumento en la liberación del hierro. El estímulo directo que controla a la hepcidina aún permanece desconocido pero parece que la HFE, la hemojuvelina y el receptor de transferrina 2 jueguen un papel importante en él. En presencia de un gen HAMP (del inglés: hepcidinantimicrobialpeptide) normal, que codifica para la proteína hepcidina, la HFE mutante puede alterar de alguna manera las señales o los factores necesarios para la síntesis de la hepcidina de los hepatocitos lo que producirá una liberación descontrolada de hierro desde los enterocitos duodenales y los macrófagos. Estos eventos explican la liberación inapropiada de hierro. (23-25)
Mutaciones en el gen HFE
Se han descrito más de 20 mutaciones en el gen HFE responsables de la hemocromatosis tipo 1, en el gráfico 3 se representa la localización de las mutaciones en la proteína. Se conoce que un 80-90 % de los pacientes con hemocromatosis hereditaria son homocigotos C282Y/C282Y; un 3,6 % heterocigotos C282Y, un 5 % son heterocigotos dobles, C282Y/H63D; un 1,5 % son homocigotos H63D y un 5,2 % heterocigotos H63D. (2,4,5)
Mutación C282Y
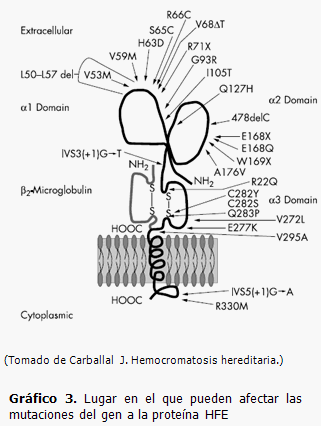
Esta mutación puntual en el aminoácido 282 (nucleótido 845 del exón 4) que cambia una cisteína en una tirosina, (26) deshace un puente disulfuro en la hélice α3, lo que provoca la eliminación de su capacidad de unión a la β2-microglobulina, y evita la expresión de la proteína HFE en la superficie de la célula. (14,15) En sistemas modelo (ratones knock-in) con la mutación C260Y (equivalente al humano C282Y), se ha demostrado que no está presente un efecto de anulación completa del alelo, ya que se produce una sobrecarga férrica menor que en ratones sin el gen HFE (ratones knock-out). Además, la sobrecarga no está presente en el momento del nacimiento, indicando que esta mutación provoca un aumento de absorción del hierro a nivel intestinal y no afecta a la captación placentaria de hierro. (27) Sin embargo, la localización de la proteína HFE y del receptor de la transferrina en la membrana placentaria, parecería indicar alguna función en la homeostasis materno-filial del hierro sugiriendo una posible participación en algunas formas de sobrecarga férrica neonatal. (28)
El estudio de la prevalencia de esta mutación en 42 poblaciones de distintos países, ha determinado que el alelo C282Y tiene su origen en las poblaciones de origen céltico y nórdico. En el Reino Unido e Irlanda el 10-20 % de la población es portadora, siendo más baja (hasta un 2-4 %) en los países del Sur y Este europeos. (27)
Penetrancia de la mutación C282Y
A pesar de los estudios realizados al respecto, la penetrancia exacta de los homocigotos para la C282Y en la hemocromatosis no se conoce aún, pero las divergencias entre el número de pacientes, la frecuencia alélica de la mutación C282Y en la población general, y las predicciones de la frecuencia de la hemocromatosis hereditaria indican que, o bien la penetrancia de la mutación es incompleta o que existen muchos pacientes no diagnosticados. (26,29,30) Seis grandes estudios poblacionales describen un porcentaje de entre el 12 al 81 % de individuos con la mutación C282Y en homocigosis con valores normales de ferritina sérica. (2,27,28). Esta amplia divergencia en el cálculo de la penetrancia de la mutación C282Y (18,8 a 88,4 %) puede deberse, en parte, a que los estudios de cribado normalmente describen una media de penetrancia en una población de individuos con un amplio rango de edad, en lugar de analizar solo el grupo de individuos de la tercera edad, ya que la expresión de la hemocromatosis depende de la edad. (2,4,5,26) Ni un exceso de hierro de la dieta ni pérdidas de sangre por menstruación o por donaciones explica todos los casos de C282Y homocigotos asintomáticos, por lo que se apunta hacia que mutaciones en otros genes pudieran modificar la expresión fenotípica de la enfermedad. Es posible que otros factores o mutaciones además de la C282Y sean necesarios para la aparición de la enfermedad, por lo que no todos los individuos C282Y homocigotos con una bioquímica alterada desarrollarían síntomas clínicos, como se apuntó en el último trabajo realizado sobre la penetrancia de la mutación C282Y en la población de EE.UU. (25,31)
Mutación H63D
La mutación H63D resulta de un cambio puntual en el nucleótido 187 (187 C → G) del exón 2 del gen HFE, que provoca en la transducción la sustitución en el aminoácido 63 de una histidina por ácido aspártico. (5) Las moléculas portadoras de la mutación H63D se procesan correctamente y se expresan en la membrana, uniéndose al receptor de la transferrina, y al parecer no alteran la afinidad del receptor por la transferrina. (2,4,5)
Esta mutación tiene una distribución más global que C282Y, lo que indicaría un origen anterior. Las frecuencias alélicas más elevadas se localizan en Europa, Asia Menor y la India, es muy poco frecuente en África y América. España con un 25-30 % de portadores es uno de los países con mayor frecuencia de esta mutación.
Penetrancia del genotipo heterocigoto compuesto C282Y/H63D
Se ha descrito que la penetrancia del genotipo heterocigoto compuesto es muy baja ya que el equilibrio Hardy-Weinberg predice en la población más heterocigotos compuestos que C282Y homocigotos y sólo un bajo porciento de heterocigotos compuestos presentan la enfermedad. Esta penetrancia ha sido estimada entre 0,44 %-1,5 % por varios grupos. (2,11)
Estudios moleculares
Los estudios moleculares en las familias con hemocromatosis hereditaria se iniciaron en 1996 utilizando la reacción en cadena de la polimeriza o PCR (del inglés: polymerasechainreaction) seguida de digestión enzimática. (11) Esto supuso un avance importante para las familias ya que se pudo abordar la detección de portadores a partir del núcleo familiar. Actualmente con la caracterización de las mutaciones es posible confirmar el diagnóstico clínico de los pacientes con clínica sugestiva.
El análisis de las mutaciones C282Y y H63D permite un nivel de detección del 90 % al 95 % utilizando el PCR seguido de digestión enzimática (RFLP). (11) Pero además de este método, existen otros como el PCR con cebadores de secuencia específica (ASO), SSCP (del inglés: single-strand conformational polymorphism) y PCR en tiempo real. (32)
PCR seguido de digestión enzimática
Comparado con otros procedimientos este método no requiere de equipamiento especializado y es probablemente el más utilizado en los laboratorios para la detección de las dos mutaciones. En Cuba, desde el año 2009 se utiliza este método para la detección de la hemocromatosis tipo I en el Laboratorio de Biología Molecular del Centro Nacional de Genética Médica, aunque también es utilizado el ASO cuando el número de muestras es elevado.
La mutación C282Y crea una diana de restricción para la enzima Rsa I o SnaBI y la mutación H63D destruye una diana de restricción de la enzima Mbo I o BclII, por lo que los alelos normales y mutados pueden ser fácilmente reconocidos sobre la base del tamaño de los fragmentos, una vez digeridos con la enzima adecuada y sometidos a electroforesis en un gel de agarosa. Muchos laboratorios utilizan los cebadores descritos por Feder y colaboradores en el artículo que detalla el descubrimiento del gen HFE. (11) Sin embargo es importante la selección de los cebadores y la enzima de restricción para que el producto de PCR contenga un sitio de restricción adicional al creado o destruido por la mutación, para usarlo como control interno de la reacción. Así pues para mutación C282Y se utiliza la enzima Rsa I en lugar del SnaBI que es de corte menos frecuente con los cebadores descritos originalmente por Feder y colaboradores. (11) Para la mutación H63D es mejor rediseñar los cebadores para que en el producto de PCR se incluya otra diana Mbo I. Debido a la polémica sobre la utilización de los cebadores reversos descritos por Feder y colaboradores en esta técnica se ha recomendado no utilizar cebadores que comprendan polimorfismos conocidos.
CONCLUSIONES
Debido a su alta incidencia, la hemocromatosis tipo I constituye un grave problema de salud en la actualidad del cual Cuba no se encuentra exenta. Por esta razón, debido a la importancia que tiene diagnosticar esta enfermedad en la fase asintomática con el objetivo de evitar el deterioro de diversos órganos, lo que pudiera comprometer la calidad de vida de los pacientes y ocasionar elevadas pérdidas económicas a la sociedad, se considera oportuno que los facultativos conozcan más sobre este padecimiento, así como sobre los medios disponibles con que cuentan para su adecuado diagnóstico.
