INTRODUCCIÓN
Las queratodermias pertenecen al grupo de las genodematosis y son aquellas dermatopatías heterogéneas cuyas principales manifestaciones se presentan en piel y anejos; tienen como elemento común su condicionamiento genético. Se produce por trastornos de la queratinización junto a la ictiosis, pitiriasis rubra pilaris y la poroqueratosis de Mibelli. (1-4)
Estas afecciones se distinguen por un engrosamiento anormal de palmas de las manos y plantas de los pies. Se clasifican en dos grandes grupos: hereditarias y adquiridas.
Las queratodermias hereditarias incluyen:
- Queratodermias simples: con queratodermia palmo plantar (QPP) como única expresión.
- Queratodermias complejas: con QPP y lesiones asociadas en áreas cutáneas no volares como el pelo, los dientes, las uñas o las glándulas sudoríparas, incluidas las displasias ectodérmicas.
- Queratodermias sindrómicas: con QPP acompañadas de trastornos en otros órganos incluida sordera y procesos malignos.
Existe otra clasificación que las divide en:
- La QPP difusa: caracterizada por una hiperqueratosis pronunciada, regular y simétrica en toda la extensión de las palmas y plantas. Por lo general esta variante es evidente al nacer o se manifiesta durante los primeros meses de la vida.
- La QPP focal: caracterizada por el desarrollo de masas compactas y voluminosas de queratina en áreas de fricción recurrente y sobre todo en las plantas de los pies.
- La QPP punteada: caracterizada por múltiples queratosis en lágrimas que pueden afectar toda la superficie palmo plantar o presentar una distribución más restringida de las lesiones (Ej. pliegues palmares).
Las queratodermias simples difusas comprenden QPP:
- QPP epidermolítica difusa (Vorner).
- QPP no epidermolítica difusa (Unna Thost).
- QPP transgredients: enfermedad de Meleda. (1-5)
La queratodermia palmo plantar de Unna Thost o queratodermia ortoqueratósica difusa como también se le conoce, es una de las queratodermias hereditarias que con mayor frecuencia se presenta en la práctica dermatológica. La incidencia es de un caso cada 40 000 personas. Se hereda como un trastorno autosómico dominante. Aparece durante los dos primeros años del nacimiento y perdura toda la vida. (1,2,5)
Clínicamente, la enfermedad se caracteriza por una queratodermia bien delimitada, simétrica y a menudo cerúlea que involucra a toda la región palmo plantar y se interrumpe bruscamente en el nivel de las muñecas, con una banda eritematosa en la periferia. (6-9)
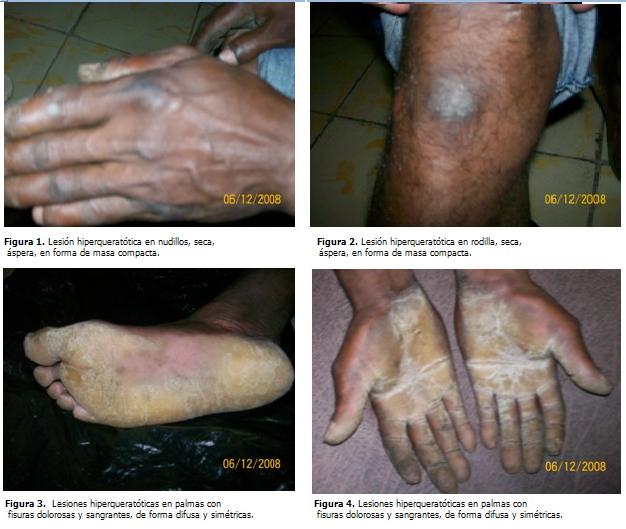
La propagación de la hiperqueratosis hacia el dorso de las manos y de las muñecas es un rasgo variable en la mayoría de las familias. A menudo la enfermedad afecta el dorso de las manos con un engrosamiento símil- esclerodermia de la piel distal con respecto a la articulación intrefalángica proximal. Hay frecuente hiperhidrosis y dolor en las zonas que se fisuran. No hay cambios asociados en el cabello y/o dientes. (10)
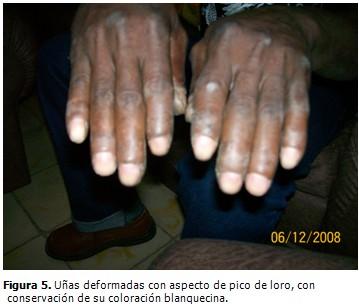
El aspecto en pico de loro de las uñas de las manos y el ensanchamiento de la banda onicorneana son hallazgos característicos de esta enfermedad. Se puede observar una hiperqueratosis de los nudillos en empedrado, pero el trastorno afecta rara vez los codos y las rodillas.
La infección secundaria por dermatofitos, con maceración y exfoliación de palmas y plantas, es una complicación frecuente de esta variante de queratodermia. (6,11)
La histología de este patrón muestra hiperqueratosis marcada, con acantosis y papilomatosis, se diferencia de la QPP epidermolítica por la ausencia de epidermólisis. La evaluación histológica de miembros de la familia original con la enfermedad de Unna Thost originó dudas acerca de la existencia real de este patrón de QPP y llevó a investigadores posteriores a considerar que esta enfermedad es una identidad clínica independiente. (12,13)
En su tratamiento se deben tener en cuenta:
- Consejos genéticos.
- Tratamiento tópico: solo producen una ligera mejoría de la hiperqueratosis. Se utilizan sustancias queratolíticas con o sin oclusión, como:
- Ácido salicílico al 5-10 % en solución acuosa de proplenglicol al 30 %.
- Ácido láctico al 20-30 % en solución acuosa.
- Urea al 10-12 % en petrolato.
En presencia de una dermatofitosis coexistente es eficaz un tratamiento prolongado con un antimicótico sistémico; por ejemplo, el uso del itraconazol en dosis de 100 mg al día. La pomada de Whitfield es un agente queratolítico tópico eficaz.
Para el tratamiento sistémico se emplean los derivados retinoicos como el etretinate de 1-1,5 mg /Kg. /día.
Son enfermedades crónicas que no mejoran con el transcurso de la vida, en algunas formas clínicas los pacientes quedan inutilizados. (1-5, 8 ,11-13)
PRESENTACIÓN DEL CASO
Se presenta el caso de un paciente de color de piel negra, masculino, de 37 años de edad, jubilado, que desde los 2 años de edad, según refirió, la madre comenzó a notarle aspereza en las manos. Durante sus estudios en el Politécnico Forestal de Pinar del Río presentó un incremento de los síntomas que fue interpretado como alergia a las plantas. Posteriormente trabajó en la refinería de petróleo donde manifestó incapacidad para realizar sus activididades laborales por lo que acudió a la consulta de dermatología de su área de salud.
En los antecedentes patológicos familiares se recogió: madre, hermana y hermano materno con discreta hiperqueratosis, tres tíos y cuatro tías por vía materna con lesiones similares, las cuales desaparecieron al arribar a la tercera edad. Sus tres hijos no presentaban lesiones.
A la exploración física se observó lo siguiente:
La piel presentaba lesiones hiperqueratósicas en palmas y plantas, con fisuras dolorosas y sangrantes de forma difusa y simétrica, rodeadas de halo eritematoso. Lesión hiperqueratósica seca, áspera en forma de masa compacta en rodillas y nudillos. (Figuras 1 a 4).
Uñas deformadas con aspecto en pico de loro, con conservación de su aspecto blanco. Ensanchamiento de repliegues supraungulares. (Figura 5).
No apareció ninguna otra alteración cutánea.
Exámenes complementarios:
Se le realizó biopsia de piel. El examen histológico reveló hiperqueratosis marcada, acantosis y papilomatosis con ausencia de epidermólisis de los queratinocitos de la capa basal y granulosa compatible con queratodermia ortoqueratósica de Unna -Thost.
CONCLUSIONES
Después de corroborar la clínica con el estudio histopatológico se pudo concluir que el paciente es portador de una queratodermia ortoqueratósica de Unna-Thost, afección heredo –familiar que por lo infrecuente de su presentación, la afectación biológica, repercusión psicológica y limitación laboral que presupone es elemento importante a incorporar al arsenal de conocimientos de nuestros profesionales.
El paciente que ahora se presenta, se mantiene en consulta para realizar el control y seguimiento de sus manifestaciones clínicas con el adecuado apoyo emocional necesario en esta entidad dermatológica.
