INTRODUCCIÓN
En 1884, Schultze (1) señaló por primera vez la existencia de una amiotrofia que se diferenciaba del resto de las atrofias musculares. En 1886, Charcot y Marie (2) en Francia y Tooth (3) en Inglaterra reportaron una forma particular de atrofia muscular progresiva cuyo rasgo más sobresaliente era su aparición en los pies y piernas y una tendencia a afectar primero los músculos peroneales, por lo cual Tooth propuso denominarla "tipo peroneal de atrofia muscular progresiva". Déjerine y Sottas (4) reportaron un cuadro clínico similar al de los autores señalados, pero diferente por la presencia de importantes fenómenos sensitivos de tipo tabético e hipertrofia de los nervios periféricos. Dyck y Lambert, (5) analizaron los caracteres de esta entidad y la clasificaron en: A) hipertrófica, B) neuronal y C) espinal. Esta clasificación fue posteriormente modificada por ellos (6) al definir siete tipos diferentes de "neuropatías motoras y sensitivas hereditarias" (NMSH): tipo I o neuropatía hipertrófica con herencia dominante, tipo II o atrofia muscular peroneal de tipo neuronal con herencia dominante, tipo III o neuropatía hipertrófica de la infancia (enfermedad de Déjerine-Sottas) con herencia recesiva, tipo IV o enfermedad de Refsum, tipo V o NMSH con paraplejía espástica, tipo VI o NMSH con atrofia óptica y tipo VII o NMSH con retinitis pigmentosa. El tipo "espinal" no es incluido por Dyck en esta clasificación.
Existe un sistema de clasificación aceptado (7) que distingue las formas desmielinizantes de la enfermedad de Charcot-Marie-Tooth (CMT) también denominadas CMT1, de las secundarias a la degeneración axonal (CMT2); la CMT3 es la enfermedad de Déjerine-Sottas, la CMT4 representa las formas autosómicas recesivas de CMT y la CMTx hace referencia a las variantes ligadas al cromosoma X. Otra clasificación alternativa (8) dispone a las NMSH en tal forma que la tipo I corresponde a la CMT1, la NMSH II a la CMT2, la tipo III a la enfermedad de Déjerine-Sottas y la tipo IV a la enfermedad de Refsum. Además existe otra clasificación, la de Rankin y Ellard, basada en la genética, que incluye a la CMT dentro de las laminopatías. (9-14)
El corazón es comúnmente afectado por algunas enfermedades degenerativas y hereditarias como la CMT, produciendo miocardiopatía dilatada o hipertrófica, trastornos del ritmo cardiaco o disturbios de la conducción, (10) aunque hay autores (11-15) que están en contra de esta relación.
El objetivo de este trabajo es presentar el caso de un paciente portador de la enfermedad de CMT con una miocardiopatía dilatada, primero de su tipo no solo en nuestro hospital en 45 años, sino también en Cuba
PRESENTACIÓN DEL CASO
Paciente del sexo masculino, de 50 años de edad, soltero, campesino, con bajo nivel cultural, sin hábitos tóxicos, ni contacto con sustancias nocivas. Antecedentes familiares: abuelos maternos y paternos fallecidos por ancianidad, el padre falleció en un accidente, la madre de neoplasia de mama, tenía un tío vivo sin problemas de salud. El paciente no poseía descendencia. Refirió que a los 14 años comenzó a presentar dificultad en la marcha (marcha en puntillas) y en un plazo de varios años se le desviaron los pies hacia dentro y la musculatura de las extremidades inferiores comenzó a atrofiarse, sufriendo caídas al caminar, todo esto junto a sensación de frialdad con coloración rojo-azulada y dolores intensos, por lo que había acudido a los facultativos en múltiples oportunidades. Desde hace varios años había notado que los síntomas se agudizaban en los miembros inferiores y por último en los miembros superiores. Debido a estas molestias, ha tenido que emplear muletas durante mucho tiempo y desde hace dos años una silla de ruedas. El paciente refirió además que desde los 30 años presentaba disfunción sexual eréctil.
Fue hospitalizado luego de acudir al servicio de urgencias por haber sufrido mareos, disnea, cianosis, palpitaciones, desmayos a repetición y arritmias (fibriloaleteo auricular).
Examen físico
Aparato cardiorrespiratorio: frecuencia respiratoria de 28 resp/min, murmullo vesicular disminuido globalmente, crepitantes bibasales, latidos cardiacos arrítmicos y taquicárdicos por fibrilación auricular; aumento global del área cardiaca; no se auscultan soplos.
Tensión arterial: 150/80 mm Hg. Frecuencia cardiaca central: 124 lat/min; pulsos periféricos presentes y sincrónicos.
Abdomen: sin alteraciones.
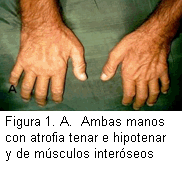
Sistema músculo-esquelético: tórax: escoliosis de concavidad izquierda. Manos: atrofia bilateral de las eminencias tenar e hipotenar y de los músculos interóseos (Figura 1A).
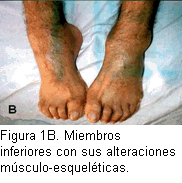
Pies: coloración rojo-azulada con moteado difuso, ausencia de vello, edemas maleolares, posición bilateral en equinovaro (Figura 1B). Raíz de nervios periféricos palpables en ambos miembros inferiores. Sistema neurológico: consciente, orientado, marcha equina. Miembros superiores: fuerza muscular segmentaria distal disminuida, con atrofia tenar e hipotenar, temblor fino bilateral de las manos tanto en reposo como al realizar movimientos voluntarios. Sensibilidad táctil y dolorosa disminuidas, con hipersensibilidad al frío. Sensibilidad profunda: normal, percibe el compás de Weber. Incoordinación en la prueba dedo-nariz. Miembros inferiores: atrofia en “calcetín”, atonía, astereognosia; sensibilidad táctil y dolorosa disminuidas, con hipersensibilidad al frío, dolor a los movimientos pasivos. Arreflexia osteotendinosa bilateral en los cuatro miembros.
Pupilas isocóricas y normorreactivas. Fondo de ojo: normal. Nistagmo horizontal espontáneo. Temblor de la lengua. Agudeza visual disminuida durante el día. La exploración de los demás pares craneales fue normal. No se constatan signos meníngeos. Exploración urológica: normal.
Estudios analíticos
Hemoglobina: 140 g/L; hematócrito: 0,48; leucocitos: 7x109/L con fórmula normal. Conteo de plaquetas, tiempos de coagulación y sangrado, enzimas hepáticas y pancreáticas, iones y función renal: todos dentro de la normalidad. Eritrosedimentación: 10 mm/1ª h. Glucemias seriadas y prueba de tolerancia oral a la glucosa: normales.
Insulina sérica en ayunas: 60 pmol/L. Colesterol total: 125 mg/dL, HDL: 37,5 mg/dL, LDL: 50 mg/dL. VDRL: no reactiva. Hierro sérico: 14 mcmol/L, ceruloplasmina: 280 mg/L, plomo: 15 mcg/dL; vitamina B12 sérica: 450 pmol/L; coproporfirina en orina: 160 mcg/24 h. CPK: 160 UI/L; isoenzimas: MM: 158 UI/L; MB: 3 mcg/L; BB: 0 UI/L. Testosterona total: 400 ng/dL, cortisol plasmático: 200 nmol/L. Estudios de líquido cefalorraquídeo: sin alteraciones. Células LE, crioglobulinas, complemento sérico, prueba del látex para factor reumatoideo, anticuerpos antinucleares, títulos de antiestreptolisina O y electroforesis de proteínas: dentro de la normalidad.
Electrocardiograma: fibriloaleteo auricular permanente con frecuencia cardiaca de 120 lpm.
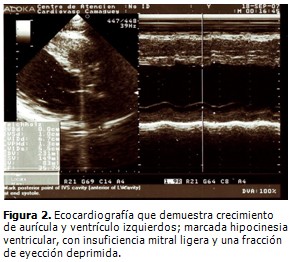
Ecocardiografía (Figura 2): ventrículo izquierdo dilatado: diámetro en diástole: 67 mm; diámetro en sístole: 55 mm; pared posterior del ventrículo izquierdo: 13 mm; aurícula izquierda: 43 mm; crecimiento de aurícula y ventrículo izquierdos; marcada hipocinesia ventricular, insuficiencia mitral ligera, no se observó derrame pericárdico, ni masas intracardiacas. Fracción de eyección deprimida (moderada): 35,7 %. Resultado compatible con miocardiopatía dilatada.
Ecografía de abdomen, próstata y tiroides: sin alteraciones.
Tomografía axial computarizada de retroperitoneo y mediastino: normales.
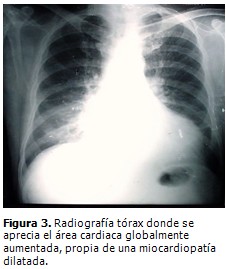
Radiografía de tórax (Figura 3): aumento global de la silueta cardiaca, con signos de congestión pulmonar.
Electromiografía: se exploraron los músculos peroneales, pedios, gemelos y tibial anterior en reposo; no se detectaron potenciales de denervación en ninguno de los músculos explorados, excepto en ambos pedios donde se hallaron ondas positivas. Al intento de contracción voluntaria se obtuviron unos trazados muy pobres de grado simple y características neurológicas. Prueba de estimulación-detección: velocidad de conducción motora (VCM) del ciático poplíteo externo izquierdo disminuida a 24,5 m/s, con potencial motor evocado simple de 3 mv de amplitud (disminuido). VCM del ciático poplíteo externo derecho de 27,4 m/s con potencial motor evocado simple de 6 mv de amplitud. Realizando estimulación del ciático poplíteo interno a nivel del hueco poplíteo, en el gemelo izquierdo se encontró un potencial motor evocado simple de 4 mv de amplitud. Los hallazgos de electromiografía resultaron compatibles con neuropatía de CMT.
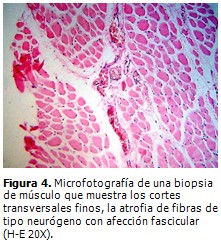
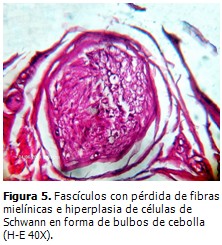
Se realizó biopsia de músculo gemelo derecho, que mostró grupos de miofibrillas en racimos atróficos mezclados con segmentos de desmielinización y remielinización (Figura 4). Biopsia de nervio ciático poplíteo interno, que demostró neuropatía desmielinizante hipertrófica con “bulbos de cebolla” como evidencia de una remielinización crónica con pérdida de fibras de mielina, preferentemente las de diámetro mayor en el curso de una NSMH tipo CMT. (Figura 5).
Tratamiento
Restricción de sal, digitálicos, diuréticos y vasodilatadores, con ostensible mejoría de las manifestaciones cardiacas; antiinflamatorios no esteroideos (ibuprofeno) para los dolores y gabapentina, 900 mg diarios vía oral, para la depresión psíquica. El paciente recibió el alta hospitalaria a los treinta días con tratamiento y fue enviado al ortopedista para implantación de prótesis ortopédica. Mantiene seguimiento en consulta de medicina interna.
DISCUSIÓN
Teniendo en cuenta el cuadro clínico, la velocidad de conducción y la histopatología, se concluyó que el paciente era portador de una enfermedad de CMT tipo 1, (13-18) si bien faltaron los estudios genéticos. Su inicio a la edad de 14 años con atrofia muscular de miembros inferiores de evolución lenta y muy pocas manifestaciones sensitivas, con trastornos de la marcha, pérdida del equilibrio, debilidad muscular y deformidades de ambos pies (19-22) (lo que ocasiona tropiezos, caídas y lesiones de tobillo) con dedos en martillo, fueron hallazgos reportados en nuestro enfermo; así mismo lo fueron la debilidad distal, la hiporreflexia osteotendinosa, la afección del sensorio, temblores, atrofia en miembros superiores e inferiores, pero más dominante y severa en los inferiores, la excesiva dureza y la palpación de los nervios, presentes en el 20 % de los enfermos. (23-27) Las malformaciones esqueléticas tales como pie cavo, genu valgus y cifoescoliosis (12, 13) también se presentaron en este caso, así como trastornos neurovegetativos en forma de cianosis, manos y pies fríos, mareos y pérdida de visión diurna, señalados por otros autores; (28, 30-32) otros hallazgos fueron la disfunción sexual eréctil desde su juventud, caída del pelo y edema de miembros inferiores, los cuales generalmente son fríos y con acrocianosis. (29)
La enfermedad de CMT es rara, (33-35) con una prevalencia de 1 caso por 2500 pacientes, según señala Skre, (17) sin embargo, Emery (18) señala un caso por cada 10.000 pacientes. Al CMT1 corresponden dos tercios de los casos y al tipo 2 un tercio, mientras que las otras formas de la enfermedad son extremadamente infrecuentes. No tiene predilección por un determinado color de la piel (32, 36, 37-39) y aunque la relación hombre-mujer no se ha determinado, en un estudio efectuado por Molina Martín et al, (15) la enfermedad afectó al sexo masculino en el 60 % y a los de piel blanca en el 86,7 %, e influyendo el factor hereditario en forma esporádica en el 66,6 % de sus pacientes; otros estudios (7, 17, 40-43) reportan que aproximadamente un 60 % de los pacientes con CMT1 son portadores de una duplicación del cromosoma 17, por lo que es muy probable que en este paciente también haya actuado el componente genético.
Además del cuadro clínico, también se corresponden con el diagnóstico de la enfermedad de CMT tipo 1, los resultados del estudio histopatológico y de las técnicas de medida de impulso nervioso y velocidad de conducción motora (que en la importante serie de Obach et al (21) de 51 enfermos se expone que pueden ser normales, algo disminuidas, bajas o muy bajas, como se presentó en este paciente).
Desde 1979, Isner et al (44) encontraron en una serie de 68 pacientes con CMT, que 29 tenían diferentes tipos de alteraciones cardiacas: en cinco se presentaron defectos de conducción; en dos, taquicardia supraventricular; en otros dos, isquemia coronaria aguda; y en 20, prolapso de la válvula mitral, debiendo realizar el diagnóstico diferencial con la ataxia de Friedreich que en muchas ocasiones puede confundirse con CMT. Últimamente, se ha señalado por algunos autores (19, 20-25, 45-47) la presencia de hipertrabeculación/compactación del ventrículo izquierdo, mientras que otros (21, 48) han demostrado enfermedad de las arterias coronarias. La miocardiopatía dilatada ha sido otra asociación comunicada en esta enfermedad, (10-12, 22, 49-52) como en el caso que nos ocupa confirmada por ecocardiografía, la cual se acompañó además de trastornos del ritmo demostrados por electrocardiograma; incluso, Sevillano Fernández et al. (53) la señala en dos hermanos. También se ha informado bloqueo auriculoventricular completo en otros estudios; (10, 24-30, 54) no obstante Dyck et al, (11) en su serie de 12 casos demuestra una fuerte evidencia en contra de la asociación de miocardiopatía y NSMH, mientras otros (25) señalan que esta asociación es controversial.
En el paciente presentado, se utilizó el tratamiento habitual para la miocardiopatía dilatada, mientras que para la CMT se emplearon los AINEs para el dolor y la gabapentina para el estado depresivo, (12) además se indicó prótesis ortopédica para los miembros inferiores, mejorando notablemente a su egreso.
Se concluye que el internista debe considerar toda la relación de aspectos clínicos expuestos y realizar un estudio simple pero sistemático de las alteraciones del músculo esquelético y los nervios periféricos, que puede incluir enzimas musculares, exámenes neurofisiológicos y biopsia de músculo, todo lo cual contribuye a aclarar diagnósticos oscuros.
