INTRODUCCIÓN
Las discapacidades del desarrollo son un grupo diverso de limitaciones físicas, cognitivas, sicológicas, sensoriales y del habla que comienzan en cualquier momento desde el inicio del desarrollo hasta los 18 años de edad. Los niños con discapacidades del desarrollo, comparados con la población general, tienen un riesgo incrementado de desarrollar epilepsia. El retraso mental (RM), la parálisis infantil (PC), el trastorno por déficit de atención con hiperactividad (TDAH) y el autismo constituyen los trastornos del desarrollo más frecuentes en la infancia. (1)
La epilepsia es uno de los trastornos neurológicos mayores más frecuente en la infancia, que afecta entre 4 y 10 niños por cada 1 000. (2-4) La prevalencia de la epilepsia en los niños con discapacidad del desarrollo es de 30 a 50 %, mucho mayor que en la población general. En la mayoría de los casos, los procesos patológicos causantes de la discapacidad del desarrollo también son responsables de la epilepsia. (1)
Como regla general, la epilepsia en niños con discapacidad del desarrollo es difícil de controlar y es más probable que se haga refractaria al tratamiento; (5) también se destaca que el riesgo de estatus epilépticos es mayor. La frecuencia de las crisis se incrementa con la severidad de la discapacidad del desarrollo.
Como parte del proyecto de colaboración en salud entre Cuba y Venezuela se realizó, en este último país, desde julio de 2007 hasta octubre de 2008, un estudio integral de personas con discapacidad del desarrollo. En dicho estudio participó un neurólogo, como parte del grupo de especialistas cubanos que laboraron en el mismo. Dicho especialista brindó asistencia especializada a través de una consulta de neurología infantil, lo cual permitió ofrecer una atención más integral a un número importante de niños y adolescentes venezolanos detectados durante el estudio.
Teniendo en cuenta la elevada prevalencia de la epilepsia en las personas con discapacidad del desarrollo, se realizó esta investigación con el objetivo de describir las principales características clínicas y terapéuticas de un grupo de niños y adolescentes discapacitados con epilepsia.
MÉTODOS
Estudio descriptivo que incluyó 364 niños y adolescentes con epilepsia, provenientes de un universo de personas con discapacidad del desarrollo, de 8 estados venezolanos en los que se llevó a cabo el “Estudio clínico-genético de personas con discapacidad”, entre julio de 2007 y octubre de 2008.
Los integrantes del equipo médico que llevó a cabo el referido estudio, remitieron a todos los niños y adolescentes con enfermedades neurológicas complejas a la consulta especializada de Neuropediatría, con el objetivo de brindar una atención más especializada.
De los pacientes atendidos en la consulta de Neuropediatría se seleccionaron los casos que cumplieran los siguientes criterios de inclusión: edad menor de 18 años al momento de la consulta y epilepsia confirmada (se consideró epiléptico a todo caso que hubiera presentado dos o más crisis epilépticas no provocadas). Se excluyeron a los pacientes con una sola crisis, un solo episodio de estatus epiléptico o múltiples crisis ocurridas en un período de 24 horas. También se excluyeron los casos con crisis neonatales o crisis febriles y los pacientes que solo presentaron crisis agudas sintomáticas (crisis asociadas con enfermedades sistémicas o neurológicas agudas, intoxicación, abuso o supresión de sustancias).
En el momento de la consulta de cada paciente se recopilaron los siguientes datos: sexo, características de la epilepsia (tipo de crisis epiléptica y síndrome epiléptico, etiología, tratamiento con drogas antiepilépticas y control de las crisis en los últimos 12 meses) y comorbilidad con discapacidades del desarrollo (RM, PC, TDAH y autismo).
Se tuvieron en cuenta para el diagnóstico y el tratamiento de los niños con epilepsia, las notas clínicas de los médicos venezolanos que habían asistido previamente a estos pacientes, así como los resultados de los estudios complementarios de electroencefalografía y de neuroimágenes (TAC y RMN de cráneo).
El diagnóstico del tipo de crisis y síndrome epiléptico se basó en las clasificaciones de la Liga Internacional Contra la Epilepsia (ILAE) de 1981 y 1989 respectivamente. (6,7) El tipo de síndrome epiléptico se clasificó según tres niveles, primero según localización (focal, generalizado e indeterminado); luego según la etiología (idiopático, sintomático y criptogénico) y finalmente en síndromes específicos.
Para el tratamiento se precisó si tomaban drogas antiepilépticas (DAE), ajustando la dosis o iniciando el tratamiento, en los casos necesarios, según el tipo de crisis o síndrome epiléptico. Se consideró controlado de la epilepsia a todo paciente sin crisis en los últimos 12 meses previos a la consulta, independientemente de si tomara o no DAE.
El retraso mental se diagnosticó con el apoyo de las pruebas psicométricas disponibles o por el juicio clínico del neurólogo. La presencia de parálisis cerebral fue precisada en la consulta a través del examen físico realizado a cada paciente.
Los datos fueron almacenados en base de datos tipo Excel 2003. Se utilizó estadística descriptiva para mostrar medidas de tendencia central, de dispersión y frecuencias. El análisis de los datos se llevó a cabo con el programa estadístico Statistical Package for Social Science (SPSS), versión 13.
Los resultados se presentan en tablas con números absolutos y porcentaje.
RESULTADOS
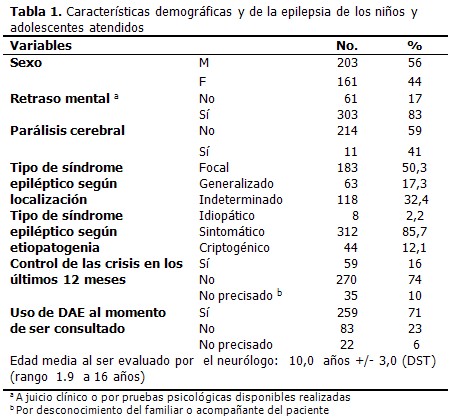
De una muestra inicial de 894 pacientes menores de 18 años atendidos en la consulta de Neuropediatría, se confirmó el diagnóstico de epilepsia en 364 casos (41 %). El 56 % son masculinos; el 83 % sufre retraso mental; el tipo de síndrome epiléptico que predominó según localización fue el focal y según etiopatogenia, el
sintomático, con 50, 3 % y 85, 7 % respectivamente. (Tabla 1).
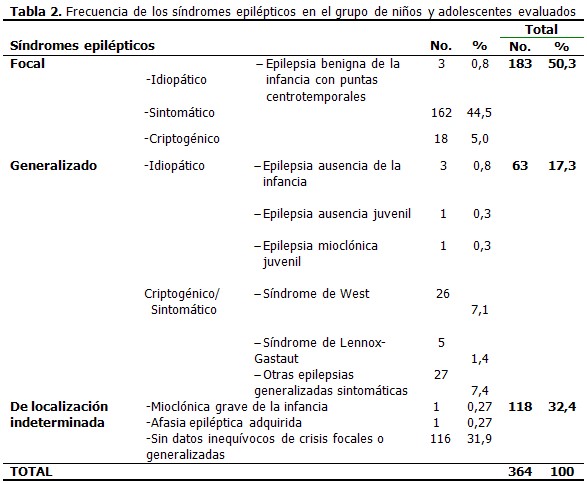
Predominó el síndrome epiléptico focal y dentro de este el sintomático; el síndrome de localización indeterminada se presentó en el 32, 4 % de los pacientes, entre los cuales el 31, 9 % se debió a la inexistencia de datos inequívocos de crisis focales o generalizadas. (Tabla 2).
De los 312 pacientes detectados con epilepsias sintomáticas, se pudo precisar la etiología responsable de la epilepsia en 105 casos: infecciones del sistema nervioso central 41; malformaciones del SNC, 39; encefalopatía hipóxico-isquémica, 16; y síndromes neurocutáneos en 9 (6 con el complejo de esclerosis tuberosa). En los pacientes con epilepsias sintomáticas que no se logró identificar la etiología específica, se determinó que eran sintomáticas, al estar asociada la epilepsia a RM y/o PC.
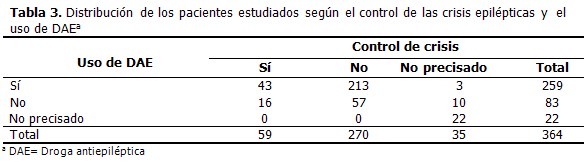
Del total de pacientes, 259 usan drogas antiepilépticas, pero sólo 43 tenían control de la crisis, mientras en 213 no existía dicho control. (Tabla 3).
Es de destacar que en los 83 casos sin tratamiento con DAE, se inició tratamiento en los pacientes con epilepsia activa (al menos una crisis en el último año). Se usó la carbamazepina (aportada gratuitamente a cada paciente) para los casos con epilepsias focales y el valproato de magnesio en los pacientes con epilepsias generalizadas. Se ajustó el tratamiento y se hicieron recomendaciones higiénico-sanitarias a 213 casos que no tenían control de las crisis pese al uso de DAE.
DISCUSIÓN
En la serie evaluada se detectó un alto porciento de pacientes con epilepsia (41 %), lo cual se corresponde con los estimados de prevalencia de la epilepsia en niños con discapacidad del desarrollo (entre 30 y 50 %), mucho mayor que en la población general. (1)
Varias discapacidades del desarrollo pueden coexistir en el mismo niño, lo cual incrementa el riesgo de padecer epilepsia. Este riesgo, en los niños con RM, es de 15 a 20 %, mayor en aquellos con discapacidad mental severa. La aparición de las crisis suele ocurrir en edades más tempranas en niños con RM que en aquellos que no lo tienen. La probabilidad de que un niño con PC desarrolle epilepsia es 5 veces mayor con respecto a los niños sin PC. (1,8-11)
En la serie de niños venezolanos estudiada, existió un claro predominio de las epilepsias o síndromes epilépticos sintomáticos, independientemente de su clasificación por localización, con un 86 % del total de niños y adolescentes atendidos. Los síndromes sintomáticos son aquellos en los cuales las crisis epilépticas se producen por una o más lesiones estructurales identificables del cerebro. Siguiendo las recomendaciones de la Comisión de la ILAE para estudios epidemiológicos sobre epilepsia, (12) se consideran con síndromes sintomáticos aquellos pacientes que presenten retraso mental o problemas motores, sin que necesariamente se haya identificado la causa de estos. La alta frecuencia en esta serie de los síndromes epilépticos sintomáticos está justificada por el hecho de la selección de los pacientes de un supuesto universo inicial de personas con discapacidad del desarrollo. Varios estudios han demostrado la estrecha relación que existe entre las epilepsias sintomáticas y las discapacidades del desarrollo, específicamente el RM y la PC. (13,14)
Dentro de las causas específicas de epilepsia detectadas, destacaron como las más frecuentes las infecciones del SNC, las malformaciones del SNC y la encefalopatía hipóxico-isquémica. Las malformaciones del SNC y dentro de ellas, las alteraciones del desarrollo cortical, han sido reportadas como una causa importante de epilepsia infantil, tal como se evidenció en esta serie. En niños y adolescentes, las lesiones congénitas, especialmente las malformaciones corticales, superan a las lesiones adquiridas del SNC (infecciones, traumas, hipóxico-isquémicas), como causas responsables de la epilepsia. (15-17)
Dentro de las causas específicas de epilepsia detectadas destacan las infecciones y malformaciones del SNC, a pesar de las posibilidades actuales de prevención de algunas de ellas a través de los programas de vacunación contra enfermedades infecciosas, como las meningoencefalitis bacterianas por meningococo y hemophilus influenzae, y la detección prenatal de malformaciones mayores del SNC a través de los estudios de ultrasonido prenatal y determinación de la alfa feto proteína.
Con respecto al tratamiento con DAE empleado en los pacientes al momento de la consulta, es llamativo el resultado de que el 23 % de la muestra estudiada no recibía ninguna DAE. Hay que tener en consideración que en la serie estudiada predominaron las epilepsias sintomáticas sin control de sus crisis epilépticas, grupo que debe ser tratado enérgicamente con DAE, tal como se procedió inmediatamente tras cada consulta médica realizada, según criterio del neurólogo basado en el tipo de epilepsia o síndrome epiléptico.
El alto número de pacientes detectados con epilepsia sin tratamiento con DAE, es una situación frecuente en muchos países en desarrollo. Esta condición se conoce como brecha de tratamiento, definida como el porciento de pacientes sin tratamiento con DAE de un total de pacientes. En estudios realizados en países de África y en Brasil, se han reportado cifras de brecha del tratamiento que van desde un 19 % en Brasil, hasta el 85 % en zonas de África, reflejo de un serio problema de salud. (18-21)
Como conclusiones del presente trabajo hay que destacar la alta frecuencia de la epilepsia en el grupo de niños y adolescentes atendidos, con un marcado predominio de las epilepsias sintomáticas y un elevado número de pacientes sin adecuado tratamiento. Venezuela es un país en desarrollo con grandes secuelas de inequidades económicas, sociales y de acceso a los servicios de salud. Los resultados detectados en este estudio son un reflejo de las consecuencias de esas desigualdades.
Los autores consideran importante desarrollar programas integrales y universales de atención a la mujer embarazada y al niño menor de 1 año, así como la implementación de un programa nacional de vacunación a toda la población infantil, todo lo cual contribuirá decisivamente a la reducción de las causas prevenibles de la epilepsia que aún persisten en sectores poblacionales venezolanos.
