INTRODUCCIÓN
La Osteogénesis Imperfecta (OI) o Huesos de Cristal entidad definida por Vrolik en 1849, constituye el síndrome osteoporótico hereditario de mayor incidencia en la infancia (1/30000). Clásicamente esta enfermedad se ha descrito principalmente en las manifestaciones clínicas secundarias a la osteopenia generalizada, sin embargo, constituye una conectivopatía con repercusión sistémica debida a defectos cuantitativos o cualitativos en la síntesis del procolageno I. (1-3)
Genotípicamente presenta una gran heterogeneidad la cual determina la variabilidad fenotípica. El espectro de la enfermedad es sumamente amplio y abarca desde una forma mortal hasta formas leves cuya definición puede ser dudosa. La clasificación más utilizada es la descrita por el Dr. Sillence en cuatro tipos (I, II, III, IV). (4-6)
El diagnóstico se realiza inicialmente con las manifestaciones clínicas y las imágenes radiográficas. La densitometría ósea, la biopsia de piel y los estudios genéticos moleculares permiten un mayor acercamiento al dictamen específico. (1, 4,6)
La OI no es una entidad frecuente en nuestro contexto, y constituye el advenimiento de un niño al Servicio de Neonatología con las manifestaciones clínicas y radiológicas características de esta entidad un motivo para realizar la presentación de este caso.
PRESENTACIÓN DEL CASO
Se presenta el caso de un recién nacido, de sexo masculino, que debido a un parto distócico por cesárea presenta dificultad respiratoria severa (Silverman 2 puntos) y fractura de humero derecho.
Antecedentes prenatales: Historia obstétrica anterior: embarazos 2, partos 2 (E2 - P2), abortos 0 (A0), tiempo de gestación 36,1 semanas, no presentó anemia, ni infecciones.
Antecedentes natales y postnatales: parto por cesárea (presentación pelviano y cesárea anterior), tiempo de rotura de membranas (TRM) 30 min, liquido amniótico meconial ++ cordón y placenta normales, peso 2300 gr. Ingresó en el Servicio de Neonatología por presentar dificultad respiratoria (Silverman 2 puntos) y fractura de humero izquierdo.
Durante su estadía en el Servicio tuvo una evolución tórpida con necesidad de oxigenoterapia, reanimación cardio-pulmonar (RCP) y ventilación durante toda la etapa de recién nacido, requirió múltiples tratamientos con antimicrobianos por presentar infecciones comnatales, asociadas a la ventilación que se mantuvo durante 28 días, después de eliminada la ventilación se decide realizar traqueostomía. Evolutivamente se constataron varias fracturas, todas de huesos largos, a pesar de haber realizado una manipulación adecuada.
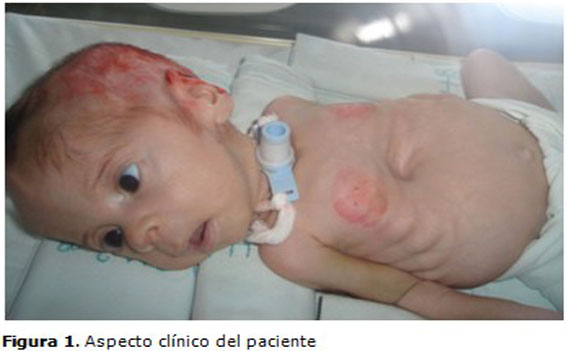
Datos positivos al examen físico: cráneo redondeado y fascie de aspecto triangular, escleras ligeramente azules que luego se tornaron blancas, excoriaciones extensas de la piel del cuero cabelludo, ausencia de surcos y pliegues palmares, hipotonía muscular e hiperlaxitud ligamentosa en manos y pies, pectus excavatum y costillas arrosariadas, piernas en abducción formando un ángulo recto con el cuerpo (postura de rana), fracturas diafisiarias de ambos húmeros y fémur derecho. (Figura 1).
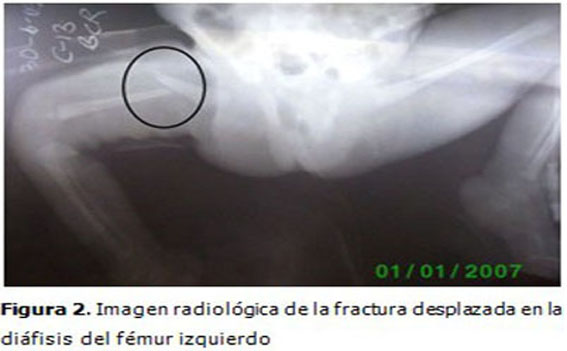
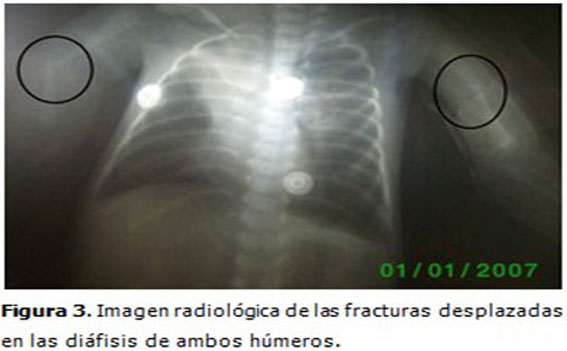
Complementarios: Se realizaron estudios radiológicos en los que se observaron osteopenia en los huesos largos y en la columna vertebral. Fracturas diafisiarias desplazadas del fémur derecho y ambos húmeros. (Figuras 2 y 3).
DISCUSIÓN
Existe un consenso general para el diagnóstico de la OI, el cual se basa fundamentalmente en la herencia, las características clínicas e imagenologícas y los estudios genéticos moleculares. Estos hallazgos permiten en gran medida tipificar la enfermedad según los criterios de Sillence, casos estos que constituyen un reto para los clínicos, ortopédicos, radiólogos y genetistas por la heterogeneidad de las expresiones. (1, 2, 4,5)
Categorizar adecuadamente los pacientes en el periodo neonatal es aún más difícil por no estar presentes totalmente las características diferenciales en cuanto a los rasgos clínicos distintivos (color de las escleras, dentinogénesis imperfecta, hipoacusia, deformidades óseas secundarias a las fracturas y displasia de tejidos blandos, etc.), sobre todo cuando se presentan como casos nuevos por mutaciones espontáneas. (4,5)
La OI tipo I, considerada la forma clínica menos grave de la enfermedad, se expresa generalmente de forma tardía en la cual se observa la clásica triada de Van der Hoeve que se manifiesta con: fragilidad ósea, escleróticas azules y sordera. La dentinogénesis imperfecta puede o no estar presente, característica que permite clasificarla a su vez en dos subtipos el A y B. (2, 6,7)
La OI tipo II se caracteriza por presentar las escleróticas gris azulado oscuro, el cráneo en pelota de goma, micromelia, fracturas óseas graves al nacer, aspecto arrosariado del tórax, deformidades óseas congénitas en las extremidades y aspecto radiológico de huesos arrugados. Su patrón de herencia es generalmente autosómico recesivo, aunque puede expresarse en mutaciones espontáneas (autosómicas dominantes). Los pacientes con esta forma clínica generalmente fallecen al nacer o a las pocas horas debido a complicaciones respiratorias, neurológicas o cardiovasculares. (4, 5, 8,9)
La tipo III se considera la forma progresiva deformante no mortal de la enfermedad, se hereda con carácter autosómico recesivo y dominante (mutaciones nuevas), los huesos generalmente son menos frágiles que en la tipo II a pesar de lo cual se pueden observar las fracturas desde el nacimiento pero con menos frecuencia y severidad. Presentan facie triangular característica y graves deformidades del tórax (pectus excavatum y costillas arrosariadas). Las escleras aunque suelen ser azules al nacer generalmente se tornan blancas posteriormente. Se acompaña siempre de dentinogénesis imperfecta e hipoacusia en la juventud. (4, 5,7)
La OI tipo IV se hereda con carácter autosómico dominante similar al tipo I pero con escleróticas normales. Estos pacientes nacen con fracturas e incurvaciones de los huesos largos de los miembros inferiores, muestran dentinogénesis imperfecta, escleróticas blancas y no hay sordera. Suele presentar cifoescoliosis y laxitud ligamentosa. (1, 4, 5,7)
Este paciente comparte rasgos clínicos y radiológicos, que hablan a favor de una OI tipo III, sin descartar una tipo IV por lo que se hace necesario evaluar la evolución futura, la cual permitirá enfocar el diagnóstico hacia un tipo específico de la enfermedad.
