INTRODUCCIÒN
El síndrome de Axenfeld-Rieger fue descrito en 1920 por Axenfeld como anomalía de Axenfeld y en 1934 ya Rieger lo describe acompañado de anomalías del iris y alteraciones no oculares como alteraciones dentarias y faciales.(1)
Es una enfermedad de origen genético, autosómica dominante en el 75 %, de alta penetrancia y expresividad variable que tienen en común su origen en la cresta neural durante el desarrollo embrionario y fetal donde se presentan mutaciones en los factores de trascripción de los genes PITX2 (4q25) y FOXC1(6p25. (2,3)Tiene una prevalencia estimada de 1/200.000.(4)
Sus manifestaciones clínicas son muy variables. Los rasgos pueden dividirse en hallazgos oculares y no oculares. Las anomalías oculares afectan principalmente al iris: hipoplasia, corectopía o formación de agujeros en el iris imitando una policoria; a la córnea: desplazamiento prominente y anterior de la línea de Schwalbe (embriotoxón posterior).
Puede estar asociado a una hipertensión ocular secundaria en el 50 % de los casos, manifestándose como un glaucoma juvenil. La disgenesia ocular puede incrementar la presión ocular (IOP) por la aparición de un embriotoxón posterior y bandas que van de la periferia del iris al embriotoxón posterior con adherencias a los procesos periféricos del iris, creando puentes de unión iridocorneales.(4-6)
Los hallazgos no oculares más característicos son: dismorfismo craneofacial leve, anomalías dentales y piel periumbilical redundante. Las anomalías del tercio medio facial incluyen: hipertelorismo, telecanto, hipoplasia maxilar con aplanamiento del tercio medio facial, frente prominente y puente nasal ancho y aplastado.(7,9)
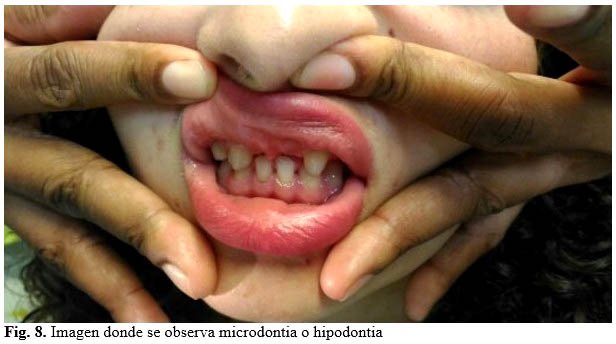
Las anomalías dentales pueden incluir microdontia o hipodontia.(1,9) También puede observarse hipospadias en hombres, estenosis anal, anomalías óseas y articulares: osificación irregular de la cabeza femoral y de las epífisis, anomalías cardiacas: defecto de los tabiques interauricular e interventricular, anomalías del sistema nervioso central: hipoacusia (disminución de la capacidad auditiva, pérdida del oído) neurosensorial, hidrocefalia (acumulación de líquido en el encéfalo), calcificaciones leptomeníngeas y retraso mental leve. Otras alteraciones menos frecuentes son deficiencia de la hormona del crecimiento y talla corta.(10,11)
El diagnóstico se basa en la combinación de los síntomas, estudios de imagen del segmento anterior del ojo principalmente la biomicroscopía ultrasónica (UBM) y la tomografía de coherencia óptica.(6)
El diagnóstico diferencial incluye: hipoplasia del iris (IH), glaucoma congénito primario (PCG), el síndrome iridocorneal endotelial, la anomalía de Peters, la distrofia polimorfa posterior, displasia oculodentodigital, iridogoniodisgenesia, aniridia, coloboma de iris y ectopia lentis et pupilae, entre otros.(9, 11)
Cada día crece más el interés por el conocimiento de las enfermedades de causa genética, existiendo muchos avances hoy en día sobre estos temas. A propósito, se expone el caso de una paciente portadora de un síndrome de Axenfeld - Rieger con antecedentes familiares de la enfermedad, portadora además de un glaucoma secundario por su enfermedad. Además, esta es una entidad cuya prevalencia es muy baja. Existen 2 casos en la Isla de Fogo, Cabo Verde.
PRESENTACIÒN DEL CASO
Paciente de 14 años de edad, sexo femenino. Natural de la Isla de Fogo, Cabo Verde. Nacida de parto normal, ligero retraso en el desarrollo psicomotor (según la madre demoró en hablar y en caminar), notó que presentaba problemas auditivos (no oye bien) pero no ha tenido estudio ni tratamiento para el mismo.
Antecedentes patológicos familiares:
Madre: no ve bien de ambos ojos y al estudiarla se comprobó que es portadora de un astigmatismo miòpico elevado en ambos ojos, ambliopía binocular y portadora del síndrome de Axenfeld – Rieger.
La paciente también refiere mala visión de lejos de ambos ojos. Al examen físico se comprobó:
Agudeza visual sin cristales (s/c) OD: cd a 3 ½ metros.
OI: cd a 5 metros.
Examen físico ocular por biomicroscopía. (Fig. 1 y 2).
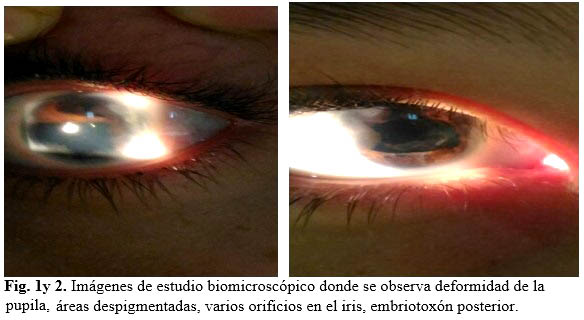
Anexos (A): ligero hipertelorismo.
Segmento anterior (S/A): en ambos ojos hay deformidad de la pupila, esta se encuentra desplazada, de aspecto atrófico, con áreas despigmentadas, presencia de varios orificios en el iris, cámara anterior desigual en profundidad, hay zonas donde el iris se encuentra adosado al endotelio corneal (sinequias anteriores), prominencia blanquecina perilímbica que se corresponde con un embriotoxón posterior.
Medios: reflejo rojo naranja de fondo (RRNF) está presente en ambos ojos.
Fondo de ojo: se entra con dioptrías negativas de +- 7 dp OD y 9 dp en OI, en el OD el disco óptico está más pálido en comparación al disco del OI, bordes bien definidos, excavación central de +- 0,4 décima, en el OD hacia el sector nasal a ½ DP se visualiza área de atrofia retiniana de forma ovalada.
Estudio gonioscópico: se visualizan todas las estructuras angulares, presencia de goniosinequias gruesas en porción superior e inferior.
Tonometría de aire: OD: 27,3 mm de hg y OI: 29,4 mm de hg.
Estudio refractivo: (VAP).
OD: - 5, 50 esfera – 6,50 cilindro x 180° (0,3).
OI: - 9,00 esfera – 2,25 cilindro x 170° (0,4)
Queratometría (Fig. 3).
OD: 38,25/ 43,75………D: - 5,50 x 5°
OI: 39,75 / 42, 75………D: -3,00 x 174 °.

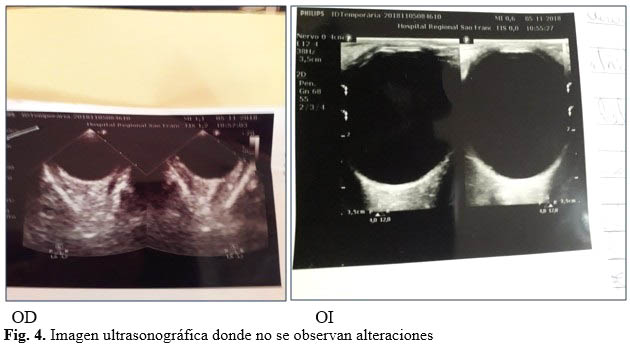
UTS ocular: no se visualizan alteraciones. (Fig. 4).
Biometría por aplanación : (Fig. 5).

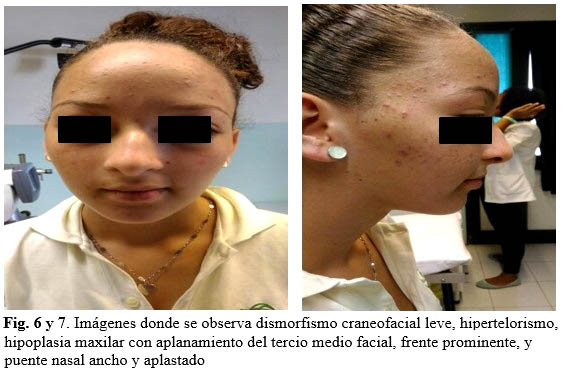
Alteraciones no oculares: dismorfismo craneofacial leve, hipertelorismo, hipoplasia maxilar con aplanamiento del tercio medio facial, frente prominente, y puente nasal ancho y aplastado. Las anomalías dentales incluyen microdontia o hipodontia. (Fig. 6, 7 y 8).
Se le indicaron cristales para corregir el defecto refractivo que tiene la paciente (astigmatismo miòpico compuesto elevado) y se comenzó el tratamiento de la hipertensión ocular con Beta-bloqueantes (timolol + dorzolamida) a dosis de 1 gota cada 12 horas combinado con los análogos de las prostaglandinas: latanoprost 1 gota en la noche. Se realizaron seguimientos periódicos en consulta.
DISCUSIÒN
En el área de las investigaciones médicas cada día se le da más valor al estudio y conocimientos de sobre las enfermedades cuyo origen tienen una base genética, existiendo hoy en día muchos avances respecto a este tema.
El caso que se expone, portador del sìndrome de Axenfeld Rieger, pertenece al género femenino con el antecedente familiar (en este caso su madre) de padecer también esta enfermedad. La paciente está en la etapa de la adolescencia, cuando comenzó a referir mala visión que se hizo acompañar de molestias oculares, ligera fotofobia y en ocasiones los ojos se le ponen algo rojos, no había sido diagnosticada anteriormente pues los familiares no la habían traído nunca al Oftalmólogo.
Al examen físico aparecieron signos referidos en la literatura revisada con afectación ocular como son las alteraciones a nivel del segmento anterior: deformidad de la pupila, esta se encuentra desplazada, de aspecto atrófico, con áreas despigmentadas, presencia de varios orificios en el iris, cámara anterior desigual en profundidad, hay zonas donde el iris se encuentra adosado al endotelio corneal (sinequias anteriores), prominencia blanquecina perilímbica que se corresponde con un embriotoxón posterior y la presencia de otros signos con afectación no ocular: dismorfismo craneofacial leve, hipertelorismo, hipoplasia maxilar con aplanamiento del tercio medio facial, frente prominente, y puente nasal ancho y aplastado así como anomalías dentales como son la microdontia o hipodontia y anomalías del sistema nervioso central: hipoacusia (disminución de la capacidad auditiva).
Ya en estos momentos la paciente presenta complicaciones propias de la evolución de la entidad como es el glaucoma secundario dado por todas las malformaciones a nivel del segmento anterior y del ángulo camerular que coexisten.
En la conducta a seguir se le realizó el estudio refractivo correspondiente donde se le indicaron sus cristales para corregir el defecto refractivo que tiene (astigmatismo miòpico compuesto elevado) y se comenzó el tratamiento de la hipertensión ocular con Beta-bloqueantes: timolol + dorzolamida 1 gota cada 12 horas combinado con los análogos de las prostaglandinas: latanoprost 1 gota en la noche con seguimientos periódicos en consulta.
En la literatura revisada también se plantea como opción la realización de una cirugía filtrante, para disminuir la presión intraocular elevada.
Los casos con glaucoma plantean un difícil reto terapéutico. Exceptuando los casos infantiles, debe intentarse la terapia médica antes de cualquier cirugía. En pacientes pediátricos el abordaje terapéutico inicial es de tipo quirúrgico, ofreciéndose la goniotomía o la trabeculotomía como tratamiento con poca efectividad. La trabeculectomía es el procedimiento quirúrgico de elección y sus resultados son comparables a los alcanzados con otras formas de glaucoma que comprometen a pacientes en un rango de edad similar. Desafortunadamente, como muchos de ellos son niños, tienen un pronóstico más reservado respecto a la utilidad de esta cirugía, por la respuesta fibrótica exagerada característica de su grupo de edad. La trabeculoplastia láser no está indicada pues es técnicamente difícil por la presencia de las adhesiones iridocorneales, así como por la inserción alta del iris, con un incremento en el riesgo de formación de sinequias anteriores periféricas que empeorarían el cuadro. La cirugía láser también se ha empleado para minimizar la corectopía.(1,12)
En el caso de esta paciente el único proceder que se ha utilizado es la triple terapia con los hipotensores oculares, se está a la espera de la respuesta al tratamiento para, en el caso de una evolución tórpida, proceder al tratamiento quirúrgico: trabeculectomía.
Siempre que se sospeche el síndrome de Axenfeld-Rieger debe evaluarse a los familiares, buscando antecedentes familiares, en este caso por un médico genetista y un médico oftalmólogo. Hay que hacerle saber a los familiares que se trata de una enfermedad autosómica dominante para tomar las precauciones necesarias.
En el caso que presentamos el estudio genético no se ha podido realizar pues el país donde vive la paciente (Cabo Verde) no cuenta con un especialista en Genética.
Conflicto de intereses:
Los autores plantean que no poseen conflicto de intereses.
Contribuciones de los autores:
Dr. Armando Rafael Milanés Armengol: atención y seguimiento del caso; búsqueda de información, redacción.
Dra. Kattia Molina Castellanos: búsqueda de información, análisis del caso, redacción.
Dra. Yusnavy Lozano Curbelo: búsqueda de información, análisis de la información, bibliografía.
Est. Marla Milanés Molina: trabajo con las referencias, trabajo con las imágenes, corrección ortográfica.
Est. Ángel Miguel Ojeda Leal: trabajo con las referencias, trabajo con las imágenes, corrección ortográfica.
Financiación:
Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos.
