INTRODUCCIÓN
Se define como anomalía de las arterias coronarias a cualquier patrón anatómico que se aparte de lo normal en lo referente a su origen, número, distribución y trayecto.
Constituyen un pequeño pero interesante grupo de malformaciones que pueden presentarse de manera aislada o en asociación con anomalías estructurales cardiacas. (1, 2)
La incidencia de las anomalías coronarias es del 2,2 % del conjunto de todas las cardiopatías congénitas (CC). (3) Las estadísticas basadas en poblaciones referidas para coronariografía encuentran anomalías coronarias entre 1-5 % de dicha población. En necropsia se encuentran hasta en el 0,3 %. (3,4) Aunque la mayoría de las anomalías coronarias congénitas aisladas son hallazgos incidentales, puede existir un riesgo elevado de dolor torácico, muerte súbita, miocardiopatía, síncope, disnea, infarto de miocardio y fibrilación ventricular; (5) como causas de ellos han sido propuestos los siguientes mecanismos: compresión de la arteria coronaria izquierda entre el tracto de salida pulmonar y la aorta, retorcimiento de la arteria a medida que se origina de la aorta, compresión súbita del orificio de la arteria anómala por expansión de la aorta y de la pulmonar durante el ejercicio, espasmo y trombosis de la coronaria izquierda en el paso entre la pulmonar y la aorta. (6)
La evaluación complementaria, en caso de sospecha de una anomalía coronaria, incluye además de la prueba de esfuerzo, otros estudios en tórax y esófago, como electrocardiograma, telecardiograma y ecocardiograma; ante su sospecha también es imprescindible la arteriografía coronaria. La resonancia nuclear magnética es de utilidad para evaluar el origen de las coronarias sin necesidad de la inyección de contrastes o la radiación, aunque la información que aporta no abarca el seguimiento del curso de la arteria, por lo que esta técnica no es de gran ayuda para determinar la presencia de fístulas o el origen de una coronaria fuera de los senos de Valsalva o de los vasos colaterales. (1, 7)
La visualización de la anatomía de las arterias coronarias es especialmente importante en la evaluación preoperatoria de los pacientes con una CC como ocurre en la tetralogía de Fallot (TF) y la transposición de grandes vasos (TGV), donde las anomalías coronarias son comunes y quirúrgicamente importantes. (8)
Teniendo en cuenta que el riesgo de isquemia miocárdica, insuficiencia cardiaca congestiva y muerte súbita, en algunos de los subtipos anatómicos, parece ser más elevado durante la niñez y adolescencia, puede asegurarse que este es un tema de interés para pediatras y cardiólogos pediatras, lo que motivó la realización de esta revisión, precisamente con el objetivo de divulgar el conocimiento relacionado con la clasificación detallada de estas anomalías.
DESARROLLO
Morfofisiología de las arterias coronarias:
Se denomina coronarias a estas arterias porque, como una corona, rodean la base del corazón; son las arterias principales de la circulación cardiaca y transportan la sangre oxigenada desde la porción inicial dilatada de la aorta ascendente (bulbo aórtico), hasta las paredes del corazón.
Tienen su origen en los senos de la aorta situados al nivel de las valvas semilunares derechas e izquierdas, (9) de manera que, durante la sístole ventricular las valvas semilunares abren el orificio aórtico, pero cierran los orificios de las arterias coronarias, ocurriendo lo contrario durante la diástole. Están situadas en el surco coronario y se clasifican como arterias de tipo muscular. Tienen un diámetro aproximado entre 5 a 6 mm en el adulto.
La arteria coronaria derecha (fig. 1) se inicia en el seno aórtico de Valsalva derecho (seno coronariano derecho), tiene un trayecto transversal más largo que el de la izquierda; se extiende por la parte derecha del surco coronario contorneando el borde derecho del corazón, pasa de la cara esternocostal a la diafragmática, donde emite la rama interventricular posterior (descendente posterior) que se continua por el surco del mismo nombre.
Su territorio de irrigación comprende la aurícula derecha (AD), la mayor parte del ventrículo derecho (VD), excepto la parte izquierda de la pared anterior próxima al tabique y una pequeña porción (parte derecha de la pared posterior que está próxima al tabique) del ventrículo izquierdo (VI). Irriga además el tabique interatrial y el tercio posterior del tabique interventricular.
La arteria coronaria izquierda (Fig. 1) se inicia en el seno aórtico de Valsalva izquierdo (seno coronariano izquierdo), tiene un trayecto corto por el surco coronario, situado por detrás del tronco de la arteria pulmonar (TAP), donde se bifurca en sus ramas interventricular anterior (descendente anterior) y circunfleja. La rama interventricular anterior desciende por el surco del mismo nombre y se anastomosa con la interventricular posterior a la derecha del ápex del corazón, la rama circunfleja se extiende por la parte izquierda del surco coronario y contorneando la cara pulmonar del corazón pasa de la cara esternocostal a la diafragmática, donde se anastomosa con la porción terminal de la coronaria derecha.
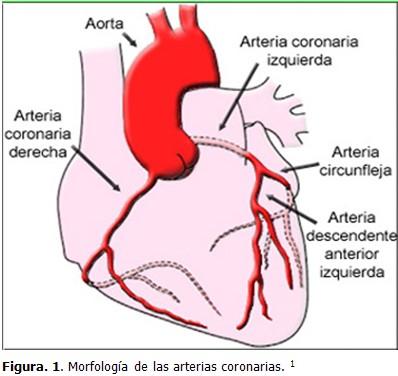
Su territorio de irrigación comprende la aurícula izquierda (AI), la mayor parte del VI (excepto la parte derecha de su pared posterior) y una pequeña parte del VD (parte izquierda de la pared anterior). Irriga además los dos tercios anteriores del tabique.
Además de este sistema de arterias que circulan en la superficie del corazón, existe un complejo entramado de arterias y arteriolas que trascurren por el interior de la pared y por el territorio subendocárdico.
En el origen embriológico de las arterias coronarias están implicados múltiples factores moleculares y bioquímicos, en un complicado proceso de migración y transformación celular, por lo que cualquier alteración que se produzca en ellos conlleva a una anomalía de las arterias coronarias, las cuales se forman entre los días 41 y 48 del desarrollo fetal.
En un inicio, las células miocárdicas en desarrollo se nutren directamente de la circulación sanguínea que proviene de la cavidad ventricular. A medida que el miocardio crece y se desarrolla surgen múltiples trabéculas las cuales originan después un sistema sinusoidal que disminuye la distancia de perfusión entre el miocito y la circulación sanguínea.
Es muy probable que de estas sinusoides se originen las arterias coronarias.
Estudios recientes han planteado el origen epicárdico del sistema vascular coronario, el cual se desarrolla de una protrusión celular del bazo primordial; estas células se establecen en las células proepicárdicas y epicárdicas y migran sobre la superficie exterior del corazón. Las células epicárdicas invaden la matriz subepicárdica y forman el plexo coronario de donde comienza a nacer una serie de vasos capilares. Cuando estos vasos están formados comienzan a extenderse y fusionarse por la cara externa del corazón, penetrando hacia el interior de este y hacia la aorta en forma de yemas que se fusionan posteriormente con los senos aórticos, originando las arterias coronarias. (10)
Entre las hipótesis que explican las anomalías coronarias, pueden citarse las siguientes: (7)
- La anómala disposición del septo aortopulmonar.
- La yema que origina las arterias coronarias se localiza de forma incorrecta en el territorio de la arteria pulmonar.
- La posibilidad de que aorta y arteria pulmonar tengan capacidad para generar las yemas de las que se forman las arterias coronarias.
El corazón es un órgano aeróbico que depende de un aporte continuo de oxígeno (O2), por lo que la función de la circulación coronaria es aportar un flujo sanguíneo siempre adecuado a las necesidades energéticas del miocardio, que varía según estas necesidades. El flujo sanguíneo del corazón se calcula entre 225 y 250 ml/min, lo que supone entre el 4-5 % del gasto cardiaco, si se asume que el peso del corazón es de unos 280 grs., esto equivale a un flujo de 70 a 90 ml/100gr/min y a un consumo de O2 de entre 8 -10 ml/100 gr. /min. La sangre fluye por las arterias coronarias a favor del gradiente. En el paciente sano, las arterias coronarias epicárdicas son vasos de conducción, al no ofrecer en la práctica resistencia al flujo sanguíneo. El flujo coronario puede estar reducido de forma fisiológica por anomalías congénitas, por razones físicas, o por situaciones patológicas, como son placas de ateromas o trombos internos en las arterias. (11)
A diferencia de otros flujos sanguíneos, el coronario fluctúa mucho a lo largo del ciclo cardiaco: durante la contracción isométrica disminuye casi hasta anularse; durante la eyección, por aumento de la presión aórtica, reaparece en menor cuantía y en la relajación isométrica es máximo, para decrecer al acercarse la nueva contracción. Por tanto, la mayor parte del flujo coronario ocurre en diástole, ya que en sístole las arteriolas coronarias intramiocárdicas se ven comprimidas por el miocardio.
A pesar de esto el flujo coronario no es semejante para ambos ventrículos, en el VI el flujo ocurre sobre todo en diástole, mientras que en el VD es más constante, teniendo lugar en sístole y diástole. Ello se debe:
- A que la presión ventricular derecha es menor, lo que favorece al flujo.
- A que las paredes del VD son de menor grosor que las del VI y más pobres en arteriolas intramiocárdicas, por lo que sufren menos la compresión miocárdica sistólica.
- A que las porciones subendocárdicas del VD se nutren directamente de la sangre contenida en el ventrículo.
El corazón es el órgano que más oxígeno extrae de la sangre, y su capacidad extractora trabaja casi al máximo incluso en condiciones basales. Por tanto la complejidad de la fisiología del flujo coronario estriba en los muchos factores que intervienen para que esa capacidad de variación sea efectiva.
El flujo coronario, como cualquier otro, es directamente proporcional a la presión de perfusión e inversamente proporcional a las resistencias coronarias. Esto se expresa por el equivalente hidráulico de la ley de Ohm. (12)
Flujo = Presión de perfusión/Resistencias coronarias.
La presión de perfusión coronaria es el resultado de la diferencia entre la presión arterial diastólica y la presión telediastólica del VI. La resistencia procede casi en su totalidad del sistema de arteriolas, en específico de las arteriolas intramiocárdicas, cuyo flujo está regulado por la sístole y la diástole, por la presión en las cámaras del corazón, y por el tono arterial que se modifica por el sistema nervioso simpático y parasimpático, así como por factores humorales o metabólicos.
El flujo coronario está determinado por la interacción de tres elementos:
- Presión de perfusión coronaria, donde la normalidad anatómica de las coronarias es un factor fundamental.
- La resistencia de la arteriola intramiocárdica, que regula el flujo mediante su capacidad de vasoconstricción y vasodilatación.
- El nivel de las necesidades miocárdicas de oxígeno.
El flujo coronario se incrementa de forma lineal para dar respuesta a cualquier incremento de las necesidades de oxígeno del corazón, mediante mecanismos de autorregulación que producen siempre vasodilatación coronaria. Los mecanismos de autorregulación más importantes sobre la circulación coronaria son el metabólico, el nervioso, el endotelial, los reflejos coronarios, y la circulación colateral.
Los estímulos necesarios para el desarrollo de la circulación coronaria colateral, son desconocidos. Se admite que el factor más preponderante es la hipoxia, el metabolismo anaeróbico produce factores metabólicos que causan vasodilatación en los microvasos preexistentes, y que son punto de partida para los futuros vasos de la circulación colateral. La existencia de circulación colateral supone casi siempre la existencia de una coronariopatía. (11)
Clasificación de las anomalías coronarias.
Existen coronariopatías congénitas y adquiridas, que pueden aparecer durante la infancia o la adultez.
La primera clasificación rigurosa data de 1969, cuando Ogden describió alteraciones mayores, menores y secundarias atendiendo a la anatomía pero no a la clínica; en 1986, Roberts determinó tres categorías de anomalías coronarias mayores en adultos; (13) el Comité de Cirugía Cardiaca Congénita estableció en el año 2000 una clasificación con siete grupos mayores de anomalías coronarias.
La baja incidencia y la escasa trascendencia clínica de algunos casos hacen que toda clasificación sea meramente orientadora. En la práctica, interesa identificar aquellas anomalías coronarias con posibilidad de afectación clínica, tributarias de tratamiento o que comporten variaciones en la estrategia quirúrgica. (8)
Se pueden clasificar en:
I. Congénitas.
a) En ausencia de cardiopatía estructural o cardiopatías congénitas.
b) Asociada a anomalías estructurales cardiacas o cardiopatías congénitas.
II. Adquiridas.
III. Asociadas a otras entidades cardiacas.
I. Congénitas.
a) En ausencia de cardiopatía estructural.
1. Origen de las arterias coronarias derecha e izquierda del seno coronario inapropiado.
Muchas de ellas se consideran variaciones anatómicas, a destacar por su asociación a casos de muerte súbita, sobre todo cuando el orificio izquierdo nace del seno coronario derecho. El trayecto de la coronaria transcurre entre los anillos aórtico y pulmonar, y durante el ejercicio se puede llegar a provocar una compresión del recorrido de la coronaria, resultando en una isquemia aguda e infarto secundario. El diagnóstico suele efectuarse en los estudios post mórtem. (3, 5)
2. Arteria coronaria única.
En un 40 % de los casos va asociado a otras CC tipo TF, tronco arterioso y válvula aórtica bicúspide. Puede nacer del seno derecho o izquierdo, puede no dar síntomas en ausencia de ateromas, pero se ha registrado muerte súbita en un pequeño número de casos, sobre todo cuando una rama pasa entre el anillo aórtico y el infundíbulo del VD. (1, 2)
3. Atresia coronaria/estenosis coronaria.
La total ausencia de una arteria coronaria extramural es rara y ocurre la mayoría de las veces asociada a atresia pulmonar y atresia aórtica. La estenosis o atresia del orificio coronario izquierdo es una de las más raras anomalías coronarias. Las ramas distales suelen ser normales y desarrollan colaterales de la arteria coronaria derecha. Pueden presentar síntomas a cualquier edad: muerte súbita, angina, infarto o fallo cardiaco. Se asocia a síndromes genéticos (Hurler, Williams, Friedrich), embriopatía rubiólica. (8)
4. Origen anómalo de las coronarias en la arteria pulmonar.
Puede tener varios orígenes como son: la coronaria izquierda naciendo en la arteria pulmonar, la coronaria derecha originándose en la pulmonar, ambas coronarias saliendo de la arteria pulmonar; o el origen de la rama circunfleja o de la descendente anterior de la pulmonar.
El origen anómalo de la coronaria izquierda desde el seno de Valsalva izquierdo de la arteria pulmonar (Fig. 2), es una anomalía coronaria grave que puede llevar a la muerte del paciente en el primer año de vida. Es la segunda anomalía coronaria clínicamente más frecuente. Es denominada como Síndrome de Bland-White-Garland, (14) debido a que en 1933 Bland, White y Garland describieron el síndrome completo, a través de sus experiencias con un lactante de 3 meses, que falleció a causa de esta enfermedad.
En la etapa fetal esta anomalía probablemente no produce daño alguno, ya que la arteria pulmonar y la aorta tienen la misma presión y una saturación de oxigeno similar, por lo que el flujo coronario es normal y hay una adecuada perfusión miocárdica, sin que exista estímulo alguno para la formación de colaterales.
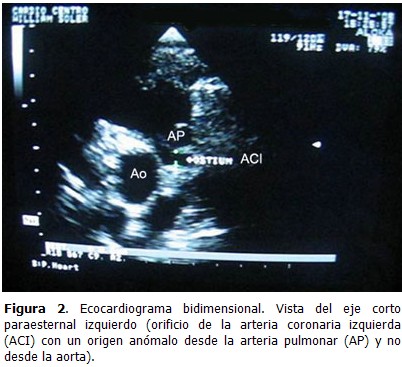
Después del nacimiento, la presión y saturación de oxígeno de la arteria pulmonar descienden rápidamente, siendo inferior a las de la aorta, por tanto, el VI (territorio de irrigación de la coronaria izquierda) que requiere de grandes demandas de oxígeno, es irrigado con sangre insaturada y con una baja presión de perfusión, lo que provoca isquemia, que en un inicio es transitoria y solo ocurre en momentos de esfuerzo (la succión y el llanto); posteriormente, con el incremento de las demandas de oxígeno del miocardio, se extiende la zona de isquemia ocurriendo un infarto en la pared anterior lateral del VI. (15)
La forma de presentación clínica se relaciona, por una parte, con la edad y, por otra, con la magnitud de la circulación coronaria colateral que comunica a las arterias coronarias derecha e izquierda entre sí. En forma genérica, se ha clasificado en cuatro grandes grupos de acuerdo con el modo de presentación o cuadro clínico a esta anomalía, lo que a grandes rasgos, concuerda con la escasa o la adecuada magnitud de la red colateral.
En las primeras semanas de vida el niño se encuentra asintomático; luego van apareciendo algunos síntomas: dificultades para la alimentación, sudoración fría, palidez, fascia de angustia, llanto inexplicable y, ocasionalmente, trastornos del estado de consciencia. Con el paso del tiempo, se establece la disnea de esfuerzo, que podrá mejorar cuando se desarrolle la circulación colateral a partir de la coronaria derecha.
Otros pacientes tendrán un cuadro dominante de insuficiencia mitral, con un soplo pansitólico apical suave e impulsos sistólicos anormales en la palpación del precordio. Se podrán presentar además signos de insuficiencia cardiaca, como son: taquicardia basal, ritmo de galope, sudoración, frialdad, pulsos débiles y rápidos, oliguria, disnea, tos, sibilancias y crepitantes, cianosis, hepatomegalia, entre otros. Ocasionalmente se apreciará un soplo continuo en mesocardio generado por las colaterales. (16, 17)
El tratamiento médico esta indicado cuando existen síntomas de insuficiencia cardiaca o disfunción ventricular, insuficiencia mitral e isquemia miocárdica. Desde el año 1933 se han descrito varias técnicas para su tratamiento quirúrgico, pero en la actualidad se realiza el reimplante directo de la coronaria anómala en la aorta. (18)
5. Fístulas coronarias.
Es una anomalía caracterizada por una comunicación anormal entre una arteria coronaria y una cámara cardiaca, arteria pulmonar, seno coronario o venas pulmonares. Pueden ser congénitas (la mayor parte de los casos hallados), o adquiridas como complicación de algún procedimiento invasivo.
Es la primera anomalía coronaria clínicamente más frecuente. Fueron descritas por vez primera en 1886 y la primera corrección quirúrgica exitosa se llevó a cabo en 1947. (19)
Desde el punto de vista embriológico, estas fístulas parecen representar uniones persistentes de la circulación sinusoidal miocárdica primitiva con los vasos epicárdicos primordiales.
Más del 50 % se origina en la coronaria derecha y un 90 % desemboca en estructuras cardiacas derechas en general, con mayor frecuencia en el VD, que suele ser el sitio de drenaje. (19, 20) Pueden ser pequeñas o dilatadas y sinuosas, con uno o varios sitios de drenaje. Las pequeñas no tienen repercusión hemodinámica, pero las grandes provocan sobrecarga de volumen.
La sintomatología está directamente relacionada con la magnitud del cortocircuito o con el tiempo de evolución de la malformación, así como a la aparición de dilataciones aneurismáticas de las fístulas que determinan mayor calibre, con el consiguiente aumento del volumen sanguíneo a través del mismo. La mayoría de las fístulas son pequeñas y los pacientes pueden presentar únicamente un soplo en el precordio; si la fístula es importante, se pueden esperar signos de fallo cardiaco predominante, pero si el flujo es pequeño los signos de isquemia son los más prominentes.
En pacientes asintomáticos se puede descubrir en un examen de rutina la presencia de un soplo de carácter continuo, que resulta del gradiente de presión entre los sistemas arterial y venoso, con derivación persistente durante todo el ciclo cardiaco. El soplo continuo de las fístulas coronarias se ausculta mejor en el segundo espacio intercostal derecho, en el área pulmonar, irradiado a la clavícula y transmitido hacia la axila o región escapular derecha.
El tratamiento consiste en ligar la fístula en su zona proximal, sin provocar afectación de la circulación coronaria distal, o en la zona de la cámara de drenaje. Recientemente se han descrito cierres de fístulas mediante cateterismo intervencionista, con coils u otros dispositivos percutáneos. (20, 21)
b) Asociada a anomalías estructurales cardiacas.
1. Transposición de grandes vasos.
La TGV es una cardiopatía compleja que se acompaña de una elevada mortalidad; es una CC relativamente frecuente que se encuentra aproximadamente entre el 5 y el 7 % de los pacientes que padecen de defectos congénitos del corazón. Se caracteriza por la existencia de una concordancia aurículo ventricular y una discordancia ventrículo arterial en la que la aorta sale del VD morfológico y la arteria pulmonar del VI morfológico. Las circulaciones pulmonares y sistémicas funcionan en paralelo, en lugar de hacerlo en serie.
El patrón coronario es muy importante para la corrección anatómica de esta cardiopatía, de ahí la importancia de su reconocimiento previo. (22) Cuando va asociada a una comunicación interventricular (CIV) o con disposición de vasos lado a lado (más frecuente en caso de doble salida de VD con CIV relacionada con la pulmonar, malformación llamada de Taussig-Bing), las anomalías coronarias o patrones coronarios son habituales y desfavorables para la corrección anatómica. Entre ellas, pueden citarse las siguientes: (22, 23)
- Arteria circunfleja originándose de la coronaria derecha.
- Origen coronario único derecho.
- Origen coronario único izquierdo.
- Arterias coronarias invertidas.
- Inversión de las arterias circunfleja y coronaria derecha.
2. Tetralogía de Fallot.
La TOF es una CC potencialmente crítica que en la mayoría de los casos cursa con cianosis, ausencia de cardiomegalia y grados variables de hipoperfusión pulmonar, que desde el punto de vista morfológico se comporta como una malformación cardiovascular de origen conal y consta de cuatro anomalías básicas que son:
- Estenosis pulmonar: obstrucción al flujo de sangre en el tracto de salida o infundíbulo del VD hacia el TAP y que frecuentemente se asocia a una estenosis de la válvula pulmonar o de las ramas.
- Comunicación interventricular.
- Dextroposición aórtica: la arteria aorta no sale del VI como normalmente ocurre, sino que lo hace por encima de la CIV, es decir entre el VD y el VI.
- Hipertrofia del ventrículo derecho: el VD está engrosado por aumento de la carga de trabajo.
Las arterias coronarias muestran variaciones a tener en cuenta para el tratamiento quirúrgico de esta enfermedad. (24) En un 4-5 % la arteria descendente anterior nace de la coronaria derecha, da origen a una rama izquierda que atraviesa el TAP en su cara anterior y pasa atravesando el tracto de salida del ventrículo derecho (TSVD); puede darse el caso de que una sola coronaria izquierda de origen a una rama derecha que atraviese el infundíbulo del VD. Si las arterias coronarias cruzan el TSVD, la cirugía correctora puede ser muy difícil, al tener que precisar la colocación de un conducto extracárdiaco para conseguir la conexión del VD con las ramas pulmonares.
3. Tronco arterioso.
Es una malformación congénita cardiovascular, en la cual una gran arteria emerge desde la base del corazón mediante una válvula semilunar única (troncal). Este gran vaso origina las circulaciones coronaria, pulmonar y sistémica. Las arterias coronarias emergen del vaso troncal en diversas variantes morfológicas. (25)
Si la válvula truncal tiene más de tres cúspides, las anomalías coronarias son frecuentes. En 2/3 del total de casos la arteria coronaria izquierda emerge del sector siniestro-dorsal del vaso troncal y la arteria coronaria derecha del sector diestro-ventral, en una posición similar a lo normal. No obstante, un origen variable de las arterias coronarias se observa con frecuencia. (26)
- Orificio coronario único (se ha precisado en 18 % de los pacientes).
- Aproximación cerrada de los orificios derecho e izquierdo (incluso del mismo seno de Valsalva) ha sido reportada en 10 % de los casos.
- Origen alto del orificio izquierdo (más frecuentemente que del orificio derecho).
- Origen de una arteria coronaria a expensas de una rama pulmonar.
- Anomalías en las ramificaciones de la arteria coronaria descendente posterior.
4. Hipoplasia de cavidades derechas.
La hipoplasia del VD tiene su origen en la atresia pulmonar con septo intacto (AP sin CIV) y la atresia tricúspide (AT), donde hay flujo disminuido al VD que impide su normal desarrollo. En las formas de VD hipertróficos, sin apenas cámara intraventricular, suele estar asociado a estenosis de los orificios coronarios apareciendo sinusoides coronarios en el VD que rellenan retrógradamente la circulación del VI; en dichas situaciones no es recomendable la descompresión del VD, pues provocaría isquemia miocárdica. (27, 28)
II. Adquiridas.
1. Enfermedad de Kawasaki.
La enfermedad o síndrome de Kawasaki es un proceso inflamatorio caracterizado por el desarrollo de fiebre, conjuntivitis, eritema marcado en mucosas, dermatitis maculo-papular en tórax, eritema difuso palmo-plantar que evoluciona hacia la descamación y adenopatías laterocervicales. La primera descripción de esta enfermedad fue realizada en Japón, por el Dr. Tomisaku Kawasaki, en el año 1961, afecta sobre todo a niños pequeños y es más frecuente en menores de 4 a 5 años, con una considerable variabilidad geográfica y racial.
Su etiología continúa siendo desconocida. Sin embargo, diversos aspectos clínicos y epidemiológicos sugieren una etiología infecciosa. Se ha relacionado con la presencia de anticuerpos IgM dirigidos contra las células endoteliales (Ac antiendotelio), que estaría desencadenada por una infección viral o bacteriana (con activación de superantígenos) los cuales facilitarían el reconocimiento de antígenos endoteliales y la subsiguiente formación de anticuerpos. Estos anticuerpos al reaccionar con el antígeno inducirían una arteritis coronaria y la formación de aneurismas.
Las manifestaciones cardiacas son uno de los hechos más importantes en esta enfermedad, es la causa más común de anomalías adquiridas de los vasos coronarios en edad pediátrica. Aproximadamente el 15-25 % de los niños no tratados desarrolla anomalías coronarias, incluyendo dilatación difusa y formación de aneurismas, que surgen por una progresiva fibrosis en las capas íntima, media y adventicia de la pared de estos vasos perdiéndose su elasticidad. Estos cambios son más pronunciados en la zona proximal de las coronarias, sugerido por el estrés hemodinámico. (29)
La dilatación coronaria puede detectarse a partir de los 7-10 días de iniciada la enfermedad, pero es entre la tercera y cuarta semana, cuando se produce el pico de mayor incidencia. La forma más severa de afectación coronaria es el desarrollo de aneurismas gigantes (≥8mm). Estos aneurismas tienen menos probabilidad de retroceder y son frecuentes las complicaciones (trombosis, ruptura, estenosis). El porcentaje de niños que desarrollarán aneurismas gigantes es variable, reportándose valores de entre 1 a 4 %, con una mortalidad elevada.
La dilatación y los aneurismas tienden a desaparecer entre 1 y 2 años en más del 50 % de los pacientes con aneurismas iniciales, sobre todo si eran aneurismas de diámetro inferior a 8 mm, se cree que debido a proliferación de la íntima de los vasos. Desde la introducción de las gammaglobulinas en el tratamiento precoz de esta enfermedad, a mediados de los años 80, se observó como los parámetros inflamatorios disminuían rápidamente, observando que las complicaciones coronarias prácticamente desaparecían.
2. Post-cirugía de cardiopatías congénitas.
Aquí se ubican las cardiopatías derivadas de la reimplantación coronaria tras la cirugía de la corrección anatómica de la TGV, en la cual se seccionan los grandes vasos por encima del plano valvular para conectarlos con los ventrículos concordantes: la aorta en el VI y la pulmonar en el VD con la reinserción de las arterias coronarias en la nueva aorta. Entre las anomalías que se pueden presentar, las estenosis de los orificios, las trombosis, y el hipodesarrollo de la circulación coronaria con la consecuente miocardiopatía secundaria, constituyen algunas de las secuelas de esta corrección quirúrgica. (22)
3. Post-trasplante cardiaco.
Una de las complicaciones más temidas del trasplante cardiaco es el rechazo crónico, que viene manifestado con una vasculopatía generalizada de la circulación coronaria que termina provocando fallo funcional del injerto. (30)
III. Asociadas a otras entidades cardiacas.
1. Miocardiopatía dilatada (MCD).
Ante cualquier MCD siempre se deben descartar anomalías congénitas coronarias que sean las causantes de dicha entidad, ello exige la realización de coronariografía que confirme una circulación coronaria normal en estos pacientes.
2. Miocardiopatía hipertrófica (MCH).
La MCH asimétrica suele ir asociada con estenosis anatómicas o funcionales de las ramas coronarias en la pared del tabique interventricular, (31) que son causantes del dolor recidivante de estos pacientes, en ausencia de obstrucción subaórtica.
CONCLUSIONES
Las anomalías de las arterias coronarias pueden asociarse a otras CC de origen troncoconal como son la TOF, la TGV y el tronco común. Es importante la visualización anatómica de estas arterias en la evaluación preoperatoria de los pacientes, ya que en ocasiones es lo que define el tipo de cirugía correctora que debe emplearse.
Pueden presentarse de forma aislada, constituyendo el 2,2 % de las CC. La fístula coronaria y el origen anómalo de la arteria coronaria izquierda en la arteria pulmonar son las más frecuentes de estas anomalías, con un elevado riesgo de dolor torácico, muerte súbita, miocardiopatía e infarto de miocardio desde las etapas tempranas de la vida.
