INTRODUCCIÓN
La polimiositis y dermatomiositis son las formas más frecuentes de miopatías inflamatorias idiopáticas, un grupo heterogéneo de enfermedades adquiridas y sistémicas del tejido conjuntivo que se caracterizan por los efectos clínicos y anatomopatológicos de una inflamación crónica no supurativa del músculo de causa desconocida.1, 2
La dermatomiositis es una entidad heterogénea con afectación sistémica y curso progresivo, que cursa con miopatía inflamatoria y manifestaciones cutáneas; pero también puede afectar otros órganos, como el pulmón, el corazón y el aparato digestivo.3
Catalogada como una enfermedad rara, afecta a adultos y niños, tiene distribución mundial y fue descrita por primera vez en Europa, hace más de un siglo. Con más frecuencia, la patología está asociada a algún trastorno autoinmunitario generalizado, a una conjuntivopatía o a alguna infección vírica o bacteriana identificada.1- 4
Se caracteriza por progresar con rapidez, tener remisiones comunes y afectación muscular global más que selectiva. El eritema característico que la acompaña facilita su identificación temprana; esto es diferente de lo que ocurre con la polimiositis, cuya fecha real de inicio es difícil de precisar y de manera típica los pacientes retrasan varios meses la consulta con el médico.3 -5
La dermatomiositis tiene su máxima prevalencia en la infancia (7-15 años) y en la mediana edad (30-50 años). Su incidencia anual calculada es de 5-10 por millón y la prevalencia de 50-110 por millón de habitantes. Igualmente significativo es su predominio en el sexo femenino con un aproximado de 3:1 y en ciertos grupos étnicos, como las personas de origen africano o latino, que tienen mayor riesgo de miopatías inflamatorias idiopáticas y peor pronóstico que los de descendencia europea.1, 3
La patogenia de esta enfermedad sigue siendo desconocida, aunque muchas líneas de investigación indican que la activación inmunitaria crónica en los sujetos con predisposición genética, tras la exposición a los desencadenantes ambientales, desempeña una función fundamental en la aparición de las miopatías inflamatorias idiopáticas.1, 6, 7
En el cuadro clínico predominan: pródromos imprecisos, edema, dermatitis e inflamación, la debilidad muscular progresiva y simétrica que afecta sobre todo la musculatura proximal de los miembros. Sin embargo, a pesar de que las afectaciones a menudo se concentran en el músculo esquelético, las miopatías inflamatorias idiopáticas son enfermedades sistémicas con manifestaciones extramusculares frecuentes que pueden convertirse en los aspectos más problemáticos de la enfermedad.3, 8, 9
La anamnesis médica y familiar cuidadosa, una exploración física completa y las pruebas de laboratorio, sobre todo las enzimas musculares séricas, la electromiografía y la biopsia muscular son claves para el diagnóstico.6
Por la baja prevalencia de esta la enfermedad y la novedad de su diagnóstico, se decidió la presentación de este caso.
PRESENTACION DEL CASO
Paciente de 30 años de edad, sexo femenino, raza blanca, de procedencia urbana, con antecedentes patológicos personales de haber sido operada de una Comunicación Interauricular (CIA) a los 4 años de edad sin que la intervención quirúrgica tuviera posteriores repercusiones en su salud. Acude al Cuerpo de Guardia del Hospital Dr. Gustavo Aldereguía Lima porque presentaba desde el día 29 de enero de 2017 astenia marcada que le imposibilitaba realizar sus actividades diarias, debilidad muscular y dolores musculares en músculos proximales de ambos miembros inferiores y superiores, además de lesiones pruriginosas acompañadas de sensación de quemazón en la piel de las 4 extremidades pero respetando el tronco. En esa ocasión es atendida en la consulta de febriles y se maneja como una sospecha de Zika; sin embargo no es ingresada y regresa a su hogar por llevar varios días con dicho cuadro.
El día 27 de febrero del 2017, después de transcurrido un mes de evolución, la paciente acude nuevamente a nuestro centro porque se mantenían los dolores musculares intensos en los músculos proximales de los 4 miembros, los cuales le impedían mantener las manos por encima de la cabeza, realizar cuclillas, subir escaleras, permanecer mucho tiempo de pie e inclusive peinarse. Durante el interrogatorio se recogen además antecedentes como: molestias al deglutir, malestar general, disnea a los esfuerzos medianos que desaparece con el reposo, tos escasa con expectoración blanquecina, ageusia, dolor en la región cervical, ligera pérdida de peso, dolores musculares frecuentes que se remontan a su adolescencia pero que no eran tan intensos como el actual. También se evidencia disfonía que la paciente ya había percibido pero que atribuía a su profesión (maestra de escuela primaria).
Al examen físico se documentaron como datos positivos:
- Eritema en placas pruriginosas en ambos miembros superiores y muslos.
- Xerostomía.
- Debilidad muscular y fuerza muscular disminuida en los músculos proximales con imposibilidad para peinarse, subir escaleras y realizar otras actividades diarias.
- Hiporreflexia rotular.
- Dolor espontáneo y a la palpación de músculos proximales de los 4 miembros, los cuales impresionan estar inflamados.
- Artralgia.
En el Cuerpo de guardia se le realizan exámenes complementarios, que muestran cifras muy elevadas de la CPK total (438 U/L). Por lo que ante estos hallazgos clínicos y de laboratorio se planteó entonces la impresión diagnóstica de una dermatomiositis, y se decidió su ingreso para estudio y tratamiento en la sala 11 A del Servicio de Medicina Interna con número de historia clínica: 459758.
Durante esta etapa de hospitalización se le indicaron a la paciente una serie de complementarios indispensables, así como otros necesarios para confirmar el diagnóstico y descartar otras patologías. Entre ellos podemos destacar:
- TGP: 117 U/L
- TGO: 216 U/L
- GGT: 56 U/L
- FA: 127 U/L
- LDH: 973 U/L
- CPK Total: 1250 U/ L
MB: 162 U/ L
- Factor reumatoideoNR
- Serología VDRL: no reactiva
- ECG: ritmo sinusal con microvoltaje en DIII, AVL, V1
- Ultrasonido abdominal, ginecológico y de tiroides: sin alteraciones.
- Ecocardiograma: Contractilidad disminuida hacia el TIV (postquirúrgica), contractilidad de PP compensatorio y del ápex con FEVI 70 %. Función diastólica conservada. Aorta conservada. Prolapso ligero de VMP que no provoca jet de reflujo. Válvulas normales no mixoma o elongación. Función diastólica del VI conservada. No dilataciones ni hipertrofia de cavidades
- Rayos- X de tórax y Radiografía de esófago- estómago- duodeno: sin alteraciones.
El 2 de marzo de 2017 la paciente es valorada por los especialistas de Reumatología, quienes describen nuevamente lesiones eritematosas a nivel de miembros superiores e inferiores y fuerza muscular disminuida en extremidades superiores e inferiores (contra gravedad y resistencia).A esto se suma las cifras elevadas de CPK llevan a plantear como diagnóstico una miopatía inflamatoria. Además sugieren realizar distintos estudios, entre ellos una electromiografía y una biopsia de músculo (de elección el deltoides) para posteriormente iniciar tratamiento con esteroides calculado en 1 mg/ kg/ diario.
Este mismo día, ante la permanencia de las molestias al tragar y disfonía de más menos 20 días de evolución, los especialistas en Otorrinolaringología realizan una laringoscopia indirecta, donde se evidencia una laringitis nodular sin la presencia de otras alteraciones que justifiquen el cuadro de la paciente.
Ante los hallazgos clínicos y de laboratorio tan sugestivos de una dermatomiositis, y a pesar de que aún no se habían realizado las pruebas diagnósticas definitivas, se decide iniciar el tratamiento de la paciente con Indometacina (25mg): 1 tableta diaria por vía oral y prednisona (20 mg) 3 tabletas a las 8 am por vía oral, con el propósito de mejorar su estado actual de salud y calidad de vida. Luego, el día 17 de marzo del 2017 se le agrega la azatioprina (50 mg) 1 tableta cada 12h. Posteriormente la paciente fue valorada en staffmeeting y discutido su caso con Anatomía Patológica, realizándose coordinaciones para biopsia y electromiografía.
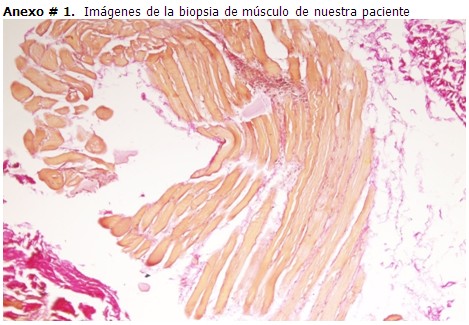
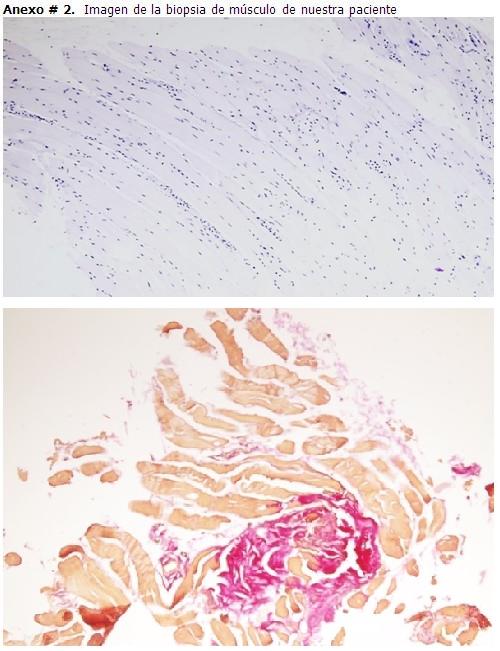
Biopsia de músculo: (B- 1795-2017) de la región deltoidea: El músculo mostró cambios en fibras aisladas consistente en núcleos sarcolémicos numerosos, con degeneración basófila del citoplasma de las fibras musculares, fagocitosis por histiocitos en numerosas fibras con cambios degenerativos aisladamente distribuidos. Al aplicar técnica tricrómica de Van Gieson se observó aumento focal del endomicio en relación con fibras musculares dañadas.
Conclusión: Patrón histológico de Distrofia muscular. (ver Anexos # 1 y 2)
Biopsia de piel: negativa.
Electromiografía: de deltoide, bíceps, oponente del pulgar, cuádriceps y gemelo. No desnervación. Patrones aislados nutrido e intermedios de 2 o´ + mv; con PUM normales. Conclusiones: EMG normal.
El equipo médico considera que los resultados de la biopsia de piel y la electromiografía estuvieron influenciados por el hecho de haber iniciado tempranamente el tratamiento de la paciente, por lo que mantiene su diagnóstico de una dermatomiositis. El 24 de marzo del 2017, al encontrarse la paciente clínicamente estable y con buen estado general, deciden el alta médica con seguimiento en Consulta Externa por Medicina Interna y Reumatología y tratamiento ambulatorio con esteroides e inmunosupresores. En la última consulta de seguimiento la paciente presentaba una mejoría notable gracias al tratamiento; sin sintomatología alguna y con cifras de CPK de 70 U/ L.
DISCUSIÓN
La dermatomiositis es una enfermedad del colágeno inflamatoria y subaguda que progresa con rapidez y tiene remisiones comunes. Su componente inmunitario lo indica: el proceso inflamatorio, la observación frecuente de autoanticuerpos y otras anomalías inmunitarias, el solapamiento con enfermedades autoinmunitarias como el lupus eritematoso sistémico en algunos pacientes, los factores de riesgo inmunogenéticos y la respuesta clínica a los fármacos antiinflamatorios.1, 5, 10
Como es el caso de otros trastornos autoinmunitarios, esta enfermedad probablemente esté provocada por una interacción entre múltiples factores de riesgo génicos y ambientales. Dentro de estos últimos, se han implicado como posibles desencadenantes de la dermatomiositis a ciertas infecciones víricas y bacterianas, así como a ciertos fármacos, citocinas, complementos dietéticos, vacunas, implantes médicos y exposiciones laborales, en casos clínicos y en algunos estudios epidemiológicos.6, 10, 11
El cambio anatomopatológico en el músculo, la piel y otros tejidos afectados se caracteriza por grupos focales de células mononucleares, además en las células musculares (miocitos) pueden mostrar también signos de necrosis, con degeneración y regeneración, con reacción fagocítica y atrofia muscular. Hay pérdida de las estriaciones transversales, cambios hialinos y granulares, núcleos picnóticos y fragmentación celular. La regeneración es típica, con cambios basófilos en el citoplasma y núcleos grandes y numerosos.En la dermatomiositis, el cambio anatomopatológico indica un proceso mediado por linfocitos B, células dendríticas y linfocitos T CD4+ cooperadores en las zonas perimisiales que hay alrededor de los fascículos y los vasos sanguíneos pequeños. Las características anatomopatológicas de los vasos sanguíneos, con lesión de la célula endotelial por el depósito de complemento, y la atrofia de las miofibras en la periferia del fascículo (llamada atrofia perifascicular) también son características de la dermatomiositis. El interferón de tipo I está presente en las células sanguíneas y en las biopsias del músculo y de la piel de la mayoría de los pacientes con dermatomiositis y en algunos pacientes con polimiositis.1, 2, 10, 12
Muchos de estos cambios antes descritos fueron apreciados en la biopsia muscular de nuestra paciente.
La dermatomiositis evoluciona de forma similar a la observada en este caso, con debilidad muscular progresiva y simétrica que afecta sobre todo la musculatura proximal de los miembros, especialmente caderas y muslos. Los pacientes presentan dificultad gradual para realizar las tareas cotidianas que requieren el uso de estos músculos como levantarse de la silla, subir escaleras, salir del coche entre otras actividades necesarias. Sin embargo, los movimientos motores finos que dependen de la fuerza de los músculos distales sólo son afectados en las fases tardías y la musculatura facial junto a los músculos oculares están respetados.4, 9, 13
En todas las formas de miopatía inflamatoria están alterados los músculos faríngeos y flexores del cuello, ocasionando disfagia, presente en nuestra paciente y dificultad para levantar la cabeza (cabeza caída); y también pueden estar afectados los músculos respiratorios pero sobre todo en los casos avanzados y rara vez en los agudos.4, 6, 11
La sensibilidad es normal en esta enfermedad y los reflejos tendinosos están preservados, aunque en los músculos con debilidad o atrofia grave pueden desaparecer. En este caso se describió al examen físico la hiporreflexia rotuliana.4
Las artralgias, por lo general simétricas, son un síntoma precoz y se observan en el 30% de los pacientes.10 Los pacientes con dermatomiositis pueden presentar además varios exantemas fotosensibles en la cara, el tórax y las manos. Las pápulas de Gottron son lesiones elevadas, a menudo descamativas y palpables sobre una base eritematosa en superficies extensoras como las articulaciones metacarpofalángicas e interfalángicas proximales, los codos, las rodillas y son prácticamente patognomónicas. Otros exantemas característicos de la dermatomiositis son el signo de Gottron (máculas en una distribución similar a la de las pápulas de Gottron) y un exantema purpúrico, característico en los párpados y a veces rodeándolos llamado exantema en heliotropo. Los pacientes pueden presentar además un exantema en la V del cuello (signo V) o en la distribución de un chal (signo del chal).1, 10, 12
Otros rasgos de la enfermedad son: el crecimiento excesivo de las cutículas, los cambios periungueales capilares y la dilatación de los capilares en las encías; este último es especialmente frecuente en la dermatomiositis juvenil. Pueden verse también: fisuras ásperas, descamativas y eritematosas en las caras palmar y lateral de los dedos, lo que se conoce como manos de mecánico.1, 10, 11
En ocasiones, los pacientes pueden presentar los exantemas clásicos de la dermatomiositis sin aparente debilidad muscular ni aumento de las enzimas musculares, en un síndrome llamado dermatomiositis sin miositis. Algunos de estos sujetos tienen, sin embargo, una miositis subclínica, como se demuestra mediante estudios de resonancia magnética del músculo o de biopsia muscular. En otros, la miositis clínica aparece con el tiempo. Los niños con dermatomiositis tienen un riesgo particular de sufrir calcificaciones subcutáneas y en ocasiones presentan una vasculitis intestinal potencialmente mortal.1, 10, 15
Es importante destacar el hecho de que a pesar de que las afectaciones a menudo se concentran en el músculo esquelético, las miopatías inflamatorias idiopáticas son enfermedades sistémicas. Entre sus manifestaciones extramusculares más frecuentes podemos destacar:
Manifestaciones generales
- Astenia
- Malestar general
- Pérdida de peso
- Fenómeno de Raynaud
- Fiebre
Manifestaciones osteomusculares
- Artralgias, generalmente simétricas que afectan a las articulaciones de la mano y artritis
- Poliarteritis no erosiva, generalmente simétrica que afecta a las articulaciones de la mano
- Artropatía deformante de las articulaciones de la mano
- Contracturas articulares
- Síndrome del túnel carpiano
- Osteopenia y osteoporosis
Manifestaciones cutáneas
- Anomalías periungueales, que incluyen telangiectasias y crecimiento cuticular excesivo
- Encías hemorrágicas por los capilares dilatados en las encías
- Placas irregulares endurecidas sobre los dedos, con acumulación de mucina en la dermis
- Calcificación subcutánea e intradérmica, que puede provocar úlceras y una infección secundaria
- Vasculitis con infartos y úlceras digitales
- Inflamación subcutánea (paniculitis)
- Alopecia
Manifestaciones digestivas
- Funciones faríngea y cricofaríngea anómalas, que suelen provocar disfonía
- Disfagia esofágica con aspiración o regurgitación nasal ocasional
- Retraso de la evacuación gástrica y reflujo
- Alteración de la motilidad de los intestinos delgado y grueso, que en ocasiones provoca dolor abdominal crónico
- Malabsorción
- Vasculitis con infartos y necrosis intestinal
Manifestaciones pulmonares
- Insuficiencia respiratoria debido a la debilidad de los músculos respiratorios
- Atelectasia
- Neumonía por aspiración
- Enfermedad pulmonar intersticial (fibrosis pulmonar)
- Hipertensión pulmonar
- Neumonitis intersticial, la cual cursa con disnea, tos y evoluciona con diferentes grados de severidad y rapidez, pruebas de función respiratoria anormales y mortalidad elevada.
- Infecciones oportunistas en pacientes inmunodeprimidos
Manifestaciones cardíacas
- Miocarditis con arritmias e insuficiencia congestiva
- El 30 % de los pacientes presentan grados variables de bloqueo, trastornos del ritmo como taquicardias auriculares y ventriculares, síndrome del seno enfermo y cambios electrocardiográficos.
- Miocardiopatía
- Cardiopatía pulmonar
- Pericarditis y derrames pericárdicos infrecuentes1-4, 9- 16
Los criterios usualmente utilizados para diagnosticar esta enfermedad, después de la exclusión de las demás formas de miopatía, fueron propuestos originalmente por Bohan y Peter en 1975 y exigen:
1. Simétrica, a menudo progresiva, debilidad muscular proximal
2. Tríada electromiografía característica:
- Potenciales polifásicos pequeños de corta duración y de baja amplitud
- Potenciales de fibrilación, se ven incluso en reposo
- Descargas anómalas repetitivas de alta frecuencia
3. Elevaciones de las actividades séricas de las enzimas asociadas a la miositis:
- Creatina-cinasa o creatinofosfocinasa (CPK)
- Aldolasa
- Lactato-deshidrogenasa
- Transaminasas: aspartato-transaminasa y alanina-transaminasa
De todas estas la enzima más sensible es la creatincinasa, la cual en la enfermedad activa puede aumentar incluso 50 veces, y cuyo valor generalmente es empleado para monitorizar la respuesta al tratamiento y la evolución de la miopatía.
4. Pruebas de inflamación crónica en las muestras de biopsia muscular:
- Fibras musculares con necrosis del tipo I y del tipo II
- Degeneración y regeneración de las miofibras, con variación en su tamaño
- Colecciones focales de células mononucleares perivasculares o intersticiales
- Eritema en heliotropo (coloración violácea de los párpados con edemaperiorbitario), dermatitis eritematosa sobre el dorso de las manos, especialmente sobre las articulaciones metacarpofalángicas e interfalángicas proximales (signo de Grotton), de la rodilla, codos, maléolo medial, cara, cuello y dorso superior.
El diagnóstico de enfermedad confirmada se logra con los cinco criterios; enfermedad probable con tres de ellos más el criterio 5 y enfermedad posible con dos de los cuatro primeros criterios, más el criterio 5 que no puede faltar nunca para confirmar el diagnóstico de dermatomiositis.1, 4, 8
En el caso específico de nuestra paciente se puede plantear una enfermedad probable, ya que el único criterio que no se evidenció fue la tríada electromiográfica.
El tratamiento de la dermatomiositis busca mejorar la potencia muscular y con ello también la función de los músculos en actividades de la vida diaria, además de reducir las manifestaciones extramusculares (disfagia, disnea, fiebre). Cuando aumenta la fuerza muscular disminuye el valor sérico de la CPK. Sin embargo, lo contrario no siempre es cierto, por lo que lo más prudente sería interrumpir estos fármacos cuando, después de un ciclo adecuado, no se observa una mejoría objetiva de la fuerza muscular, con independencia de si disminuyen o no los valores séricos de CPK. Los fármacos utilizados en el tratamiento son: glucocorticoides (prednisona) e inmunodepresores (azatioprina, metotrexato y la ciclofosfamida).4, 11 12
El pronóstico de la enfermedad se ha estudiado poco, pero la información actual señala algunos rasgos clínicos asociados a un mal pronóstico, como son: el retraso del tratamiento una vez iniciada la enfermedad, la afectación pulmonar, cardíaca o digestiva y una respuesta deficiente al tratamiento inmunosupresor. Además la dermatomiositis es considerada por algunos autores como un síndrome para neoplásico, por lo que en la medida que sea diagnosticada precozmente contribuirá al tratamiento satisfactorio del paciente.
CONCLUSIONES
La dermatomiositis es una enfermedad con una baja prevalencia y etiología que no está totalmente esclarecida. En esta paciente la evolución clínica y las pruebas complementarias nos permitieron definir el caso como una dermatomiositis e iniciar el tratamiento sistémico adecuado. La paciente evolucionó favorablemente, mejoró su sintomatología y egresó con seguimiento por consulta externa.