INTRODUCCIÓN
La incontinencia pigmenti (IP) es una enfermedad descrita por primera vez por Garrod en 1906 pero, posteriormente, fue definida por Bloch, Sulzberger y colaboradores, en función de sus características clínicas e histopatológicas, por lo que se llegó a conocer como síndrome de Bloch-Sulzberger. Tiene distribución mundial pero su prevalencia es desconocida, existiendo aproximadamente 700 casos reportados desde 1987.1
Se presenta casi exclusivamente en mujeres, puesto que casi todos los varones mueren en la etapa perinatal a menos que su cariotipo sea 47XXY o presenten una mutación hipomórfica.2,3
La causa de la incontinencia pigmenti es una mutación producida a nivel del gen NEMO/IKKg cuyo locus se encuentra en Xq28, siendo su patrón de herencia de tipo dominante ligada al cromosoma X.1,3 Los hallazgos dermatológicos son los primeros en presentarse y se dividen en cuatro estadios: vesículas -generalmente perinatales-, lesiones verrugosas, hiperpigmentación que sigue un patrón característico, y por último lesiones hipocrómicas atróficas o cicatrizales.4 Las alteraciones cutáneas pueden estar presentes en el momento del nacimiento o aparecer en los primeros 15 días de vida, con el tiempo, se van atenuando, llegando a desaparecer en la pubertad y fase adulta.
La dificultad del diagnóstico, radica en que el cuarto estadio de la enfermedad es una manifestación tardía, probablemente infradiagnosticada, que consiste en alteraciones tróficas hipopigmentadas en los miembros inferiores. En algunos casos quedan lesiones neurológicas residuales como retraso mental y/o graves afectaciones motoras y oculares.5 Los pacientes presentan fenotipo muy variable debido a la inactivación de uno de los cromosomas X (heterocromatinización) muy temprano en la embriogénesis, que determina la afección variable de distintos órganos. La afectación del sistema nervioso central y ocular pauta el pronóstico. Su detección temprana es importante ya que hoy en día no existe tratamiento etiológico, debiéndose realizar tratamiento de las diferentes manifestaciones.6
Por estas razones se decidió la presentación de este caso.
PRESENTACIÓN DEL CASO
Neonato femenina, hija de madre de 31 años de edad con pareja no consanguínea, de color de piel negra, con retraso mental ligero. La madre refirió que cuando ella nació estuvo dos meses ingresada por lesiones en piel pero no se definieron cuáles, ni se le dio un diagnóstico.
Antecendentes obstétricos: embarazo bien controlado. Parto eutócico a las 40 semanas de gestación, cefálica, peso 2 950 gramos. Recién nacido de término, peso adecuado para la edad gestacional, Apgar 9/9.
Al momento del nacimiento se observaron lesiones vesiculoverrugosas con distribución lineal en miembros inferiores y multiples máculas hiperpigmentadas de más menos 1mm, algunas más grandes distribuidas por toda la piel. (Figura 1).
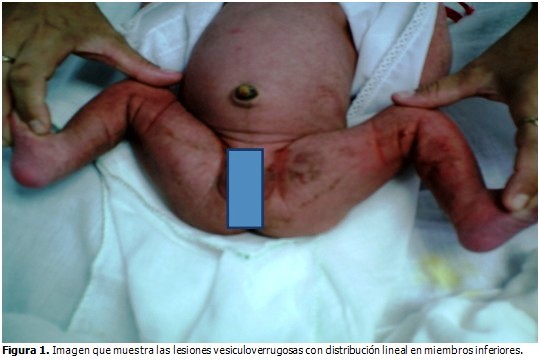
Se evaluó por el Servicio de Dermatología. Se indicaron exámenes complementarios que arrojaron los siguientes resultados:
Hemograma: Hematocrito: 0,45; leucocitos: 8 000/mm3; neutrófilos: 58 %; eosinófilos: 8 %; linfocitos: 25 %; monocitos: 8 %; basófilos: 1 %.
No fue posible realizar biopsia de las lesiones ya que la madre se negó a la realización del proceder.
Se realizó diagnóstico clínico de incontinencia pigmenti. Se indicó cura de las lesiones con loción antiséptica (Timerosal) no apareciendo nuevas lesiones. Egresó a los seis días de edad con seguimiento por la consulta de Dermatología y Genética.
DISCUSIÓN
La incontinencia pigmenti es una enfermedad genética perteneciente al grupo de las genodermatosis. Es un raro trastorno multisistémico, cuya incidencia es de 1 cada 40 000 nacidos vivos.7 Se trasmite en forma dominante ligada al cromosoma X y solo la padecen las mujeres.8 Los varones, así como las mujeres, que presentan el defecto en forma homocigota, son afectados severamente y mueren en el útero por tener el gen anormal en su único cromosoma X. La relación de afectados mujer/hombre es 20:1. A pesar de lo mencionado anteriormente, se han reportado más de 60 varones afectados. Su sobrevida se explicaría por uno de los siguientes tres mecanismos: 9,10
- Concomitancia con un cariotipo 47, XXY: correspondiente a síndrome de Klinefelter. Se estima que este mecanismo presenta el 7 % de los varones afectados.
- Mosaicismo somático: niveles bajos de mosaicismo: 46,XY/47,XXY demostrado solamente mediante la realización de FISH (Fluorescente In Situ Hybridization) en interfase usando sondas X e Y.
- Mutaciones hipomórficas que producen una forma leve de la condición (por ejemplo duplicaciones de un tracto de 7-citocina en el exon 10 del gen NEMO).
En este caso, la madre de la paciente presentaba alteraciones al examen físico y existía el antecedente de retraso mental. En un estudio realizado en 1976 con 653 pacientes se reporta un 55,4 % de historia familiar definitiva de incontinencia pigmenti.11 Esto podría explicarse porque en algunas mujeres adultas los signos cutáneos desaparecen,12 o porque las manifestaciones clínicas varían considerablemente incluso entre los miembros de una familia, producto de un mosaicismo funcional por el cual solo algunas células del mismo origen embriológico expresan la alteración genética.7
La afectación, tanto del SNC como la ocular, en el primer año de vida, ensombrecerán severamente el pronóstico de estos pacientes. Su detección precoz, por tanto, es importante, ya que hoy todavía no existe un tratamiento etiológico, debiéndose realizar un tratamiento sintomático de las diferentes alteraciones. Las complicaciones neurológicas aparecen en el 30-50 % de los casos1 y consisten en: retraso mental, crisis convulsivas, parálisis espástica, microcefalia, malformaciones cerebrales y ataxia cerebelosa. Por regla general, estos pacientes presentan un cuadro de encefalopatía aguda en época neonatal que cursa con crisis convulsivas de repetición. Las lesiones cutáneas del cuero cabelludo ubicadas principalmente en vértex en el 38 % de los casos se asocian con frecuencia a lesiones cerebrales subyacentes. Esta característica constituye una expresión de la lionización genética en estas pacientes, es decir el proceso por el cual algunas células del mismo origen embriológico expresan el defecto y otras no.13 Los ojos pueden mostrar todo tipo de alteraciones: microftalmía, papilitis, retinopatía, deformidad de los párpados, su frecuente asociación con la afectación del SNC y su posible aparición en el primer año de vida obligan a realizar controles oftalmológicos precoces. La dentición en estos niños presenta frecuentemente hipodoncia o anodoncia (en torno al 43 %), retraso en su erupción, deformidades constitucionales (dientes cónicos) y alteraciones del esmalte, siendo esta la afectación extracutánea más frecuente. Se afectan tanto los dientes temporales como los permanentes.5
A nivel óseo las alteraciones se caracterizan por la presencia de hemivértebras, escoliosis, espina bífida, sindactilia, anomalías del oído, costillas extra y deformidades del cráneo. Otra alteración característica de estos enfermos son los tumores queratósicos periungueales, que aparecen entre la pubertad y la 3ª década de la vida, localizándose sobretodo en los dedos de los pies; pueden evolucionar hacia la regresión espontánea o a veces hacia un crecimiento continuo con dolor, distrofia ungueal y destrucción ósea de la falange distal (a pesar de su histología benigna precisan, en estos casos, ser extirpados).14
El tratamiento se decide en función de las anomalías extracutáneas ya que las lesiones de piel son benignas. Las lesiones cutáneas son autorresolutivas y su tratamiento puede ser sintomático.15
Para finalizar, es importante resaltar que la IP se considera una enfermedad genética, potencialmente grave, que obliga al seguimiento multidisciplinar precoz del paciente e impone el asesoramiento genético a la familia. Por tanto, el diagnóstico precoz de estos casos asienta en el reconocimiento de las lesiones cutáneas iniciales, y así contemplar la IP como parte del diagnóstico diferencial de los eritemas vesiculoampollosos neonatales.
