INTRODUCCIÓN
El rabdomiosarcoma es un tumor maligno que se origina a partir de células musculares esqueléticas normales y como dichas células están presentes en casi todo el organismo, este tipo de tumor puede tener cualquier localización; los sitios más comunes son la cabeza y el cuello, el aparato genitourinario y los brazos o las piernas. No es muy frecuente; se diagnostican solo unos 350 casos al año en menores de 21 años en los Estados Unidos. Cada año, cuatro niños de entre un millón de niños sanos, de edad inferior a 15 años desarrollará un rabdomiosarcoma. Es ligeramente más frecuente en niños que en niñas y la incidencia máxima tiene lugar en niños pequeños por debajo de los cinco años.1-5
Los síntomas asociados al rabdomiosarcoma pueden variar ampliamente dependiendo del lugar en que este se desarrolle. Los niños con rabdomiosarcoma orbitario (alrededor del 10 % del total de los casos) pueden presentar un ojo protuyente o inflamado (proptosis), aunque puede ser confundido con una sinusitis. La mayoría de los niños que desarrollan este tipo de cáncer no tienen un factor de riesgo genético identificable.6-8 Solo entre un 10 y un 33 % de los niños que lo desarrollan sí tienen factor de riesgo genético subyacente.9
El rabdomiosarcoma es considerado como una única enfermedad, aunque existen diferencias importantes de comportamiento de este tumor en dependencia de su lugar de origen, el tipo histológico, el tamaño, su posible diseminación a otras localizaciones, la cantidad de tumor residual que queda después de la cirugía y la edad que tenga el paciente en el momento del diagnóstico, estos son lo llamados factores pronósticos. Un diagnóstico precoz de esta entidad es importante ya que es un tumor que se disemina rápidamente.10
En la práctica clínica se observa que tumores invasivos de la órbita como neuroblastoma, rabdomiosarcoma, retinoblastomas de crecimiento exofítico, pueden tener una presentación clínica inicial muy similar a la que tendrían entidades inflamatorias, infecciosas o traumáticas, por lo que se resalta la importancia de pensar en los diagnósticos diferenciales.11
La estadificación del rabdomiosarcoma infantil se realiza mediante tres formas distintas de describir el cáncer:sistema de estadificación, sistema de agrupamiento y grupo de riesgo. El sistema de estadificación se basa en el tamaño del tumor, la ubicación en el cuerpo y en si este se diseminó hasta otras partes distantes del cuerpo (se subdivide en cuatro estadios). El sistema de agrupamiento se basa en determinar si el cáncer se diseminó y si todo el cáncer se extirpó con la cirugía (se subdivide en cuatro grupos, en los que el grupo B, tiene tres divisiones). El grupo de riesgo se basa en el sistema de estadificación y en el sistema de agrupamiento (riesgo bajo, intermedio y alto).12
El Instituto Nacional del Cáncer de los Institutos Nacionales de la Salud de Estados Unidos expone que la estadificación del rabdomiosarcoma es relativamente compleja. El proceso incluye los siguientes puntos: a) asignar un estadio: determinado por el sitio primario, el tamaño del tumor en su diámetro más grande y presencia o ausencia de metástasis a ganglios linfáticos regionales o metástasis a distancia; b) asignar un grupo al tumor local: determinado por estado posquirúrgico, resecado/biopsia, con evaluación patológica del margen tumoral y enfermedad de ganglio linfático y c) asignar un grupo de riesgo: determinado por estadio, grupo, e histología. Los protocolos actuales COG-STS para el rabdomiosarcoma usan el sistema de estadificación pretratamiento con base en el TNM que incorporan al sitio de tumor primario, presencia o ausencia de invasión tumoral de los tejidos circundantes, tamaño del tumor, estado de los ganglios linfáticos regionales y la presencia o ausencia de metástasis.13
Por lo anteriormente expresado, por la importancia de su diagnóstico precoz y lo poco frecuente de la afección, se decidió la presentación del caso.
PRESENTACIÓN DEL CASO
Paciente masculino de seis años de edad, de color de piel negra, procedencia rural, sin antecedentes patológicos de interés. Acudió al Hospital General de Kuito Bie, en Angola, donde fue atendido por médicos cubanos de misión en ese país, en el año 2013.
Acudió con su padre, por presentar una masa tumoral que protuía por la cavidad orbitaria derecha.
Del interrogatorio a su padre y de los datos de la historia clínica se recogieron los siguientes elementos como antecedentes:
Fue llevado a consulta de oftalmología por su papá, debido a que presentaba inflamación y dolor en el ojo derecho, refería haber sufrido un trauma ocular pues le habían tirado una piedra una semana antes. Estuvo ingresado y se le había puesto tratamiento.
La historia clínica reflejaba en el examen físico ocular:
En la primera consulta se constató una agudeza visual de percepción de luz en el ojo derecho y 1,0 de visión en el ojo izquierdo.
Al examen con la lámpara de hendidura se observó:
Ojo derecho:
Anexos: hiperemia cilio conjuntival.
Segmento anterior: erosión corneal superficial, cámara anterior formada, humor acuoso transparente, mancha blanquecina a través del área pupilar.
Medios: reflejo rojo naranja de fondo disminuido.
Fondo de ojo: no se pudieron precisar detalles.
Ojo izquierdo: normal.
El paciente fue llevado nuevamente a consulta a los 15 días y el papá refirió que desde hacía dos días había comenzado con dolor intenso y mucha inflamación del ojo derecho.
En esa ocasión se reflejó en su historia clínica lo siguiente: al examen se encontró edema de párpados marcado, quemosis conjuntival, hiperemia cilio conjuntival intensa, córnea deslustrada, cámara anterior con presencia de hipopión de más o menos 4 milímetros que no permitía precisar otros detalles del segmento anterior. Se decidió ingresar nuevamente con tratamiento, pero al continuar evolucionando tórpidamente, se decidió realizar evisceración del ojo derecho.
El paciente, a los tres meses de la cirugía comenzó con aumento de volumen que protruía a través de la cavidad orbitaria y fue traído nuevamente a consulta, donde entonces fue atendido por los autores de este trabajo. (Figuras 1 y 2).
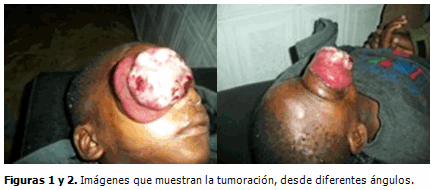
Se realizaron estudios como hemograma, parcial de orina, rayos X de tórax, todos dentro de límites normales.
Exámenes imagenológicos:
Tomografía axial computarizada de tórax y cráneo que informó: no se detectó metástasis a nivel torácico, en la de cráneo se observó imágen de aspecto tumoral de densidad variable que ocupa órbita derecha, con aumento de volumen del músculo recto externo y proptosis de 12x12 cm, así como rarefacción ósea de pared lateral de la órbita e imagen hiperdensa en región frontal del mismo lado, compatible con proceso secundario.
Se realizó biopsia de la lesión y resonancia magnética nuclear. En la resonancia se demostró presencia de una masa extensa en partes blandas que parte de la cavidad derecha.
Teniendo en cuenta que ya al paciente se le había realizado la evisceración y que según refería el padre, todo el cuadro había sido secundario a un traumatismo ocular y la tumoración comenzó al cabo de este proceso y considerando que el rabdomiosarcoma es un tumor maligno que se origina a partir de células musculares y que es más frecuente en el sexo masculino, además de observarse en niños, siempre se pensó en esta entidad. No obstante se tomó muestra del tumor, para confirmar diganóstico.
Además se pudo observar el rápido crecimiento de dicho tumor. (Figura 3) .
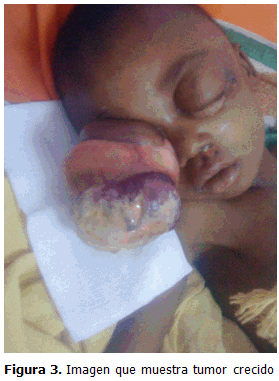
La biopsia fue positiva de un rabdomiosarcoma orbitario con variante histológica alveolar, así como metástasis.
El caso fue visto en consulta multidisciplinaria, se repitió hemograma cuyos resultados arrojaron cifras bajas de hemoglobina (8, 0 g/l).
Se concluyó como un rabdomiosarcoma orbitario con metástasis ósea y se decidió tratamiento paleativo por la toma general que ya presentaba el paciente, el cual falleció un mes después de este ingreso.
DISCUSIÓN
Las lesiones de cabeza y cuello son motivo de consulta frecuente en pediatría, la mayor parte de ellas corresponden a aumentos de volumen y generalmente con características de benignidad, se relacionan con infecciones, inflamaciones, acumulaciones de líquidos o tumefacciones. A pesar de esto, es importante considerar siempre una etiología maligna que detectada y manejada precozmente presentaría un buen pronóstico. El caso que se presenta es un ejemplo fehaciente de la importancia que tiene el diagnóstico precoz y el correcto tratamiento en estos pacientes. Este niño llegó a consulta de los médicos cubanos cuando ya se había realizado la evisceración y el tumor había alcanzado grandes proporciones.
El cáncer es la segunda causa de muerte infantil, se ha estimado que 5 - 10 % de los tumores malignos en niños se producen en cabeza y cuello y dentro de estos tumores se destaca el rabdomiosarcoma infantil que representa aproximadamente el 3,5 % de los casos de cáncer en niños de 0 – 14 años de edad.14
Es muy infrecuente la diseminación del rabdomiosarcoma a distancia, solo uno de cada cinco niños la desarrollará, por lo que es importante poder realizar estudios y evitar la diseminación de este tipo de tumor.
Todos los casos deben ser tratados con quimioterapia y muchos de ellos con combinación de radioterapia y cirugía, pero esto depende del tamaño y la localización del tumor primario y de la proporción del mismo para que pueda ser extirpada quirúrgicamente.15
Existe actualmente en el mundo la aplicación de nuevos tratamientos, entre ellos tenemos la llamada cirugía de rescate que se aplica después de lograr la reducción del tumor con la quimioterapia y se han desarrollado mucho los tratamientos basados en la biología tumoral.15
En los últimos años, el sitio predominante de fracaso del tratamiento en los pacientes con rabdomiosarcoma localizado inicialmente fue la recidiva local. Tanto la cirugía como la radioterapia son medidas principalmente tomadas para producir el control local, pero cada una tiene riesgos y beneficios. Inicialmente debe considerarse la extirpación quirúrgica de todo el tumor pero siempre y cuando esto no produzca impedimentos funcionales y cosméticos de importancia.16
Con esa condición, se recomienda la resección completa del tumor primario con un margen circundante de tejido normal junto con muestras de ganglios posiblemente comprometidos obtenidas en la órbita de drenaje linfático. Hay algunas excepciones importantes a esta regla sobre márgenes normales (por ejemplo, tumores de la órbita y de la región genitourinaria). El principio de resección amplia y completa del tumor primario se aplica menos a pacientes que tienen cáncer metastásico confirmado en el momento de la operación inicial, pero este es un concepto razonable si se puede lograr fácilmente.17
El rabdomiosarcoma representa un tercio de los tumores malignos de cabeza y cuello por lo que se debe tener presente siempre que observemos alguna masa tumoral fija, lobulada e irregular.
Las formas de presentación del rabdomiosarcoma infantil pueden confundir al médico general, retardar el diagnóstico y como consecuencia de ello comprometer el pronóstico de la enfermedad.
Se consideró de gran importancia este caso ya que en toda la literatura revisada no se encontró un tumor de este tipo que alcanzara tan grandes dimensiones en un corto período de tiempo en un niño.
