INTRODUCCIÓN
La enfermedad de Von Hippel Lindau (VHL) (OMIM 193300) es una condición genética de herencia autosómica dominante caracterizada por la aparición de quistes y masas viscerales, benignas y malignas, en múltiples localizaciones. Los pacientes pueden desarrollar hemangioblastomas del sistema nervioso central y la retina, múltiples quistes de páncreas y riñones, carcinoma renal y feocromocitomas, entre otras lesiones menos frecuentes. Esta es una de las enfermedades de predisposición hereditaria al cáncer y tiene una incidencia de alrededor de 1 en 36 000 nacimientos.1
Su etiología parte de las mutaciones germinales en el gen VHL. Este es un gen supresor tumoral localizado en el brazo corto del cromosoma 3 (3p25-26) que codifica una proteína ubiquitina ligasa involucrada en la respuesta celular a la hipoxia.2,3
La enfermedad de Von Hippel Lindau tiene una expresividad muy variable, al igual que la edad de sus primeras manifestaciones clínicas. Por ello, a pesar de existir criterios clínicos bien establecidos para su diagnóstico, la disponibilidad del estudio genético molecular de las mutaciones del gen VHL es de mucho valor. Cuando es posible identificar la mutación que causa la enfermedad en un individuo afectado, el resto de los miembros de la familia en riesgo pueden beneficiarse del diagnóstico confirmatorio y de la posibilidad de recibir asesoramiento genético.4
El diagnóstico genético molecular de mutaciones puntuales en el gen VHL requiere la combinación de estrategias de cribado y secuenciación de su región codificante, que se extiende a lo largo de tres exones.5 En Cuba la búsqueda de mutaciones germinales del gen VHL se realiza desde el año 2006 por el método de análisis de polimorfismo conformacional de simple cadena de ADN (SSCP) para los tres exones, seguido por secuenciación. Estas técnicas, aunque muy sensibles, son costosas, laboriosas y consumen mucho tiempo.
No solo para detectar y/o confirmar la presencia de una mutación puntual del gen VHL en un individuo enfermo, sino también para garantizar su fácil aplicación en el diagnóstico de otros familiares en riesgo, en especial para realizar estudios presintomáticos y prenatales, resulta más efectivo el empleo de técnicas más rápidas y económicas.5
Entre las técnicas de la biología molecular que brindan esas ventajas se encuentra la reacción en cadena de la polimerasa seguida por la digestión con enzimas de restricción (en inglés, PCR-RFLP). Esta ha demostrado ser útil para el diagnóstico de mutaciones puntuales causadas por el cambio de una base nitrogenada o pequeñas deleciones o inserciones que crean o destruyen un sitio de restricción. Se basa en la detección de fragmentos de ADN de distinto peso molecular o de longitudes diferentes, de manera que, mediante el análisis de los polimorfismos en el tamaño de los fragmentos de restricción, es posible determinar la presencia o no de la mutación, y por tanto, el genotipo del individuo.6
Este trabajo tiene como objetivo describir la introducción del diagnóstico molecular por análisis directo (PCR-RFLP) de las mutaciones c.362A>G (p.Asp121Gly) y c.481C>T (p.Arg161ter) del gen VHL, en el laboratorio de biología molecular del Centro Nacional de Genética Médica (CNGM) de Cuba.
MÉTODOS
Se realizó un estudio descriptivo de corte transversal, en el laboratorio de biología molecular del CNGM, entre los meses de enero y diciembre del año 2011. Se estudiaron pacientes con diagnóstico clínico de enfermedad de Von Hippel-Lindau y familiares en riesgo atendidos en la consulta de genética clínica del Hospital Hermanos Ameijeiras. Previo consentimiento informado, se obtuvieron muestras de sangre periférica para la obtención de ADN genómico.
Para determinar la existencia de endonucleasas cuyos sitios de restricción resultaran afectados por la presencia de las mutaciones c.362A>G y c.481C>T, se empleó el programa informático CLC Sequence Viewer, versión 6.5.1. El análisis se realizó a partir de la secuencia de ADN de referencia para el gen VHL (GeneBank: NG_008212.3).
Para la optimización y validación de la digestión enzimática se emplearon muestras de ADN pertenecientes a 24 individuos previamente caracterizados mediante secuenciación de ADN, en el laboratorio de Biología Molecular del CNGM. EL ADN se aisló mediante el método de precipitación salina,7 a partir de muestras de sangre periférica. Posteriormente se cuantificaron las muestras de ADN mediante espectrofotometría.
Para la amplificación del ADN se emplearon los cebadores: 5’-AGCCACCGGTGTGGCTCTTTA-3’ y 5’-ACATCAGGCAAAAATTGAGAACTGG-3’ para el exón 2, y 5’-CCTTGTACTGAGACCCTAGTCTGTCACT-3’ y 5’-CAAGACTCATCAGTACCATCAAAAGCTG-3’ para el exón 3 del gen VHL. Las reacciones se llevaron a cabo en volúmenes de 25 µL que contenían 0,4 µM de cada cebador; 0,1 mM de dNTPs; 2,5 U de Taq ADN Polimerasa; 1x de solución tampón de la polimerasa; 1,5 mM de MgCl2 y de 50 a 100 ng de ADN genómico. Los programas de PCR consistieron de una desnaturalización inicial a 95°C por 5 minutos, seguida de 30 ciclos de desnaturalización a 95°C por 40 segundos, alineamiento a 57°C (para el exón 2) o 54°C (para el exón 3) durante 40 segundos y polimerización a 72°C por 40 segundos, seguida de una extensión final a 72°C por 5 minutos.
La digestión enzimática para la detección de la mutación c.362A>G se realizó en un volumen de 30 µL, que contenía: 2 U de la enzima SfaN I, 1x de solución tampón de la endonucleasa y 10 µL del producto del PCR. Para la detección de la mutación c.481C>T la digestión enzimática se realizó en un volumen de 30 µL, que contenía: 2,5 U de la enzima BtgZ I, 1x de la solución tampón de la enzima, 3 µg de BSA y 15 µL del producto de PCR.
El producto de la digestión fue analizado mediante electroforesis de ADN (a 250 V por 40 minutos) en un gel de agarosa al 2 %, teñido con bromuro de etidio y fotografiado sobre un transiluminador de luz ultravioleta.
Se compararon los genotipos obtenidos mediante secuenciación y por PCR-RFLP atendiendo a las variables: presencia de la mutación c.362a>g y presencia de la mutación c.481c>t, ambas cualitativas dicotómicas.
Este artículo ha sido aprobado por el Comité de Ética de la Investigación Científica y por el Consejo Científico del CNGM.
RESULTADOS
Mediante el análisis bioinformático de la secuencia de los exones 2 y 3 del gen VHL se determinó que la mutación c.362A>G elimina el sitio de restricción de la enzima SfaN I en el segundo exón del gen. Así mismo, la mutación c.481C>T elimina el sitio de restricción de la enzima BtgZ I en el tercer exón del gen VHL.
Se estudiaron las muestras de ADN de 21 pacientes pertenecientes a dos familias con diagnóstico molecular de enfermedad de Von Hippel Lindau confirmado por la técnica de SSCP-secuenciación, además de muestras de tres individuos sanos, no portadores de ninguna de las dos mutaciones, confirmados molecularmente por la misma técnica. Entre los pacientes diagnosticados con la enfermedad, 11 tenían la mutación c.362A>G y otros 10 la mutación c.481C>T en el gen VHL.
Como resultado de la digestión del producto de PCR del exón 2 con la endonucleasa SfaN I se pudo comprobar mediante la electroforesis en gel de agarosa que en los casos diagnosticados con la mutación c.362A>G se obtenía un fragmento no digerido de 266 pares de bases correspondiente con el alelo mutado. (Figura 1).
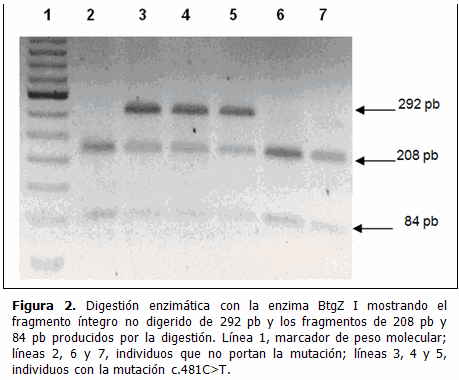
De la misma manera la digestión con la endonucleasa BtgZ I del producto de PCR del exón 3, producía un fragmento no digerido de 292 pares de bases en todos los casos con la mutación c.481C>T.
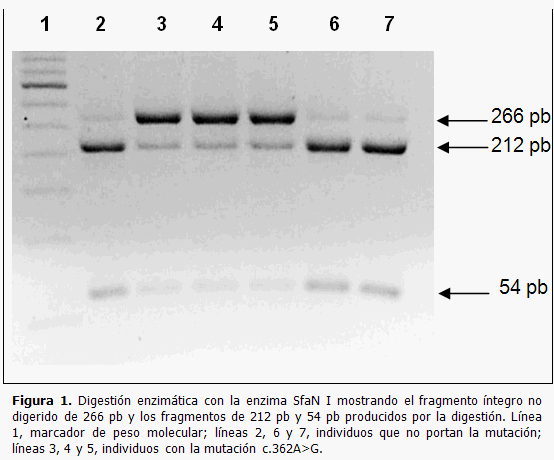
Las muestras de los individuos sanos solo muestran los fragmentos digeridos correspondientes a los alelos no mutados. (Figura 2)
DISCUSIÓN
La mutación c.481C>T (p.Arg161ter) es una de las más frecuentemente reportadas en familias afectadas por la enfermedad de Von Hippel-Lindau en todo el mundo. Así mismo, la mutación c.362A>G (p.Asp121Gly) ha sido encontrada en varias familias de ascendencia asiática y europea.8 Esta investigación presenta la introducción de una nueva estrategia para el diagnóstico molecular de estas dos mutaciones germinales del gen VHL en el laboratorio de biología molecular del CNGM, la cual se basa en la aplicación de una técnica simple y directa como la amplificación por PCR seguida por digestión con enzimas de restricción (PCR-RFLP).
Resulta destacable además, el empleo de un programa bioinformático (CLC Sequence Viewer, versión 6.5.1) para realizar el análisis de la secuencia del gen VHL e identificar las enzimas de restricción adecuadas, un ejemplo más de cómo la bioinformática puede ser exitosamente aplicada en la solución de problemas biomédicos y el valioso aporte que hace a la investigación en el área de la biología molecular.9
Mediante la estrategia propuesta se logró reproducir los resultados previamente obtenidos por SSCP-secuenciación en la totalidad de los casos estudiados. No se encontraron discrepancias entre lo obtenido con ambos métodos, pero sí se pudo constatar que la técnica PCR-RFLP es más sencilla y económica, además solo demora dos días, en comparación con los siete necesarios para completar la secuenciación. Este resultado justifica su empleo cuando se trata de buscar mutaciones ya conocidas en otros miembros de la misma familia, abriendo la posibilidad de incorporar los diagnósticos presintomático y prenatal a los protocolos de atención que reciben estos pacientes.
La técnica PCR-RFLP aunque fue desarrollada a finales de los 70, todavía se utiliza para el diagnóstico molecular de mutaciones puntuales en muchos genes causantes de enfermedad en el humano.10
En el caso del gen VHL, si bien esta técnica se usó bastante alrededor de su descubrimiento en el año 1993,11,12 las publicaciones recientes sobre el tema del estudio molecular de sus mutaciones rara vez hacen referencia a su empleo, dadas las inmensas posibilidades que brindan tecnologías más modernas y de mucho mayor alcance como la secuenciación.1,4,13
Sin embargo, en nuestro laboratorio la técnica PCR-RFLP es ampliamente empleada con resultados satisfactorios en el estudio de varias enfermedades genéticas frecuentes en Cuba.14,15 Este estudio muestra que el diagnóstico de mutaciones puntuales del gen VHL también puede ser realizado mediante su aplicación.
Podemos decir a modo de conclusión que la técnica PCR-RFLP tiene ventajas sobre la estrategia SSCP-secuenciación para establecer de forma rápida, reproducible y confiable el diagnóstico de la enfermedad VHL en casos de familias molecularmente ya caracterizadas. Esta investigación permitió estandarizar la técnica PCR–RFLP para la detección de las mutaciones c.362A>G y c.481C>T del gen VHL, lo que hace posible disponer de una estrategia simple y económica, aplicable al diagnóstico confirmatorio, presintomático y prenatal de la enfermedad de Von Hippel Lindau.
Consideramos necesario agradecer a todos aquellos profesionales: especialistas en genética clínica y másteres en asesoramiento genético, clínicos, neurocirujanos, oftalmólogos y otros, que han estado involucrados en la atención a pacientes cubanos con la enfermedad de Von Hippel Lindau. Así mismo a los trabajadores y técnicos del laboratorio de biología molecular del CNGM que han participado en la estandarización de las técnicas necesarias para el diagnóstico molecular de esta condición.
