INTRODUCCIÓN
En 1973, Cohen y colaboradores1 describen en tres pacientes un síndrome nuevo caracterizado por la asociación de obesidad, hipotonía, retraso mental, dismorfia cráneofacial típica y anomalías de manos y pies. Estas características clínicas fueron confirmadas posteriormente en cuatro casos,2 quedando establecidas las manifestaciones del síndrome de Cohen.3 Un estudio adicional en seis pacientes finlandeses añadió la distrofia coriorretiniana y la granulocitopenia asintomática al fenotipo clínico.4
Han sido muchos los síndromes familiares descritos que asocian retraso mental, obesidad, talla baja y anomalías genitales. El síndrome de Cohen, también llamado síndrome Pepper, es una entidad bien individualizada que debe diferenciarse de otros síndromes como el de Prader Willi,5 y el de Lawrence Monn Bield.6 Las anomalías cráneofaciales y el tipo de obesidad, así como el momento de aparición de la misma, permiten distinguir estos procesos.7 El síndrome de Mirhosseini-Holmes-Walton,3 caracterizado por microcefalia, retraso mental y degeneración pigmentaria tiene mucha analogía con el síndrome de Cohen, algunos autores postulan que es el mismo.7
Sin embargo, los criterios clínicos estrictos son difíciles de establecer, variando incluso en los tres casos del artículo original de Cohen.1 Existe una amplia variedad en las manifestaciones de presentación que hace sospechar que no todo se corresponde con el mismo proceso.
De acuerdo con la hipótesis enunciada se admite que el síndrome de Cohen es una afección hereditaria con transmisión autosómica recesiva, sin que se encuentren diferencias en función del sexo y con considerable variabilidad en la expresión; se han publicado varios casos en hermanos, en ocasiones con consanguinidad de los padres.8
Pocos casos hay descritos en la bibliografía cuya sintomatología se manifestase antes de los diez años de edad, lo que contrasta con nuestro estudio. Es preciso realizar un diagnóstico precoz diferenciándolos de otras entidades similares, con el fin de prevenir aquellas alteraciones, como las endocrinológicas, que aparecen en la etapa puberal.
Se publica este trabajo con el propósito de dar a conocer las características fenotípicas más comunes en estos pacientes, lo cual contribuye al diagnóstico precoz y su identificación, para poder actuar en etapas más tempranas y brindar un mejor asesoramiento genético.
Durante esta investigación se respetaron los principios éticos descritos en la declaración de Helsinki de 2010 para investigaciones médicas. Se contó con el consentimiento informado de la paciente y su mamá para la realización de los estudios requeridos y la toma de fotografías para el diagnóstico y la publicación de los resultados.
PRESENTACIÓN DEL CASO
Paciente femenina, de 14 años de edad que se atiende en el Centro Provincial de Genética Médica de Holguín desde los ocho meses de vida por hipotonía y retraso en el desarrollo psicomotor.
En la etapa preescolar también presentó retraso en la adquisición del lenguaje, dificultades en el aprendizaje, falta de memoria, déficit de atención, comportamiento anómalo y torpeza motora.
Tiene un peso mayor del percentil 97, una talla en el percentil 90 y un perímetro cefálico en el percentil inferior al tercero, por lo que presenta microcefalia y obesidad troncular. Pabellones auriculares pequeños, incisivos prominentes, micrognatia, filtro corto, raíz nasal ancha. Hendiduras palpebrales antimongoloides, manos estrechas, con dedos finos hacia la falange distal. Hipotonía de la eminencia tenar e hipotenar, pliegue palmar transverso, sindactilia cutánea parcial. Genus y cúbito valgo. Hiperextensibilidad articular. Cifosis dorsal leve e hiperlordosis lumbar. Discreta hipotonía generalizada. El cariotipo, electrocardiograma, tomografía craneal y radiografía de esqueleto resultaron normales.
Se interconsultó con Neurología, Fisiatría, Pediatría y Genética y se pensó primeramente en un síndrome de Prader-Willi. Se le indicaron periódicamente glucemias, TGP, TGO y hemogramas, los cuales siempre arrojaron resultados normales.
A la edad de cinco años fue valorada por Neurología y remitida al Centro de Diagnóstico y Orientación a la Familia por el retardo evidente del intelecto de esta niña. El grupo multidisciplinario determinó un retraso en el desarrollo intelectual de tipo moderado, por lo que comenzó los estudios en una escuela de enseñanza especial, donde aún se encuentra.
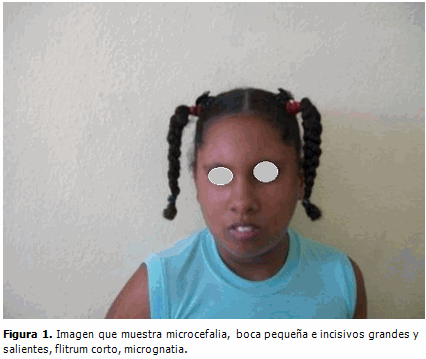
Ahora, con 14 años, son bien evidentes las características faciales que muestran microcefalia, con boca pequeña e incisivos grandes salientes, flitrum corto, micrognatia. (Figura 1).
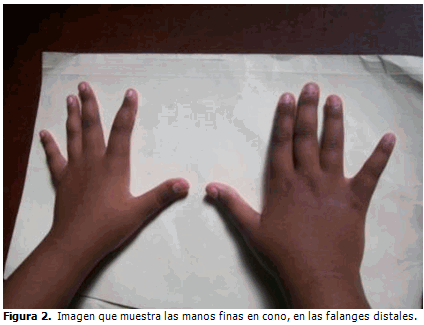
Las falanges distales de la manos las tiene más finas, con dedos en forma de huso. (Figura 2).
Llama la atención la obesidad en tronco y extremidades en la parte proximal. Sin embargo tuvo siempre un crecimiento pondoestatural acorde a su edad y sexo y en este momento tiene talla normal y la pubertad retrasada en cuanto a las características de vello y su distribución. (Figura 3).

Se determinó que la niña padece del síndrome de Cohen.
DISCUSIÓN
La mayoría de los pacientes con síndrome de Cohen tiene una alteración completa del crecimiento con talla baja, retraso en la maduración ósea, obesidad troncular y pubertad retrasada de causa desconocida.1-4
En esta niña, en que se mantuvo el crecimiento normal, sí fue evidente el retraso de la edad ósea por lo que llevó consulta por endocrinología.
La variabilidad fenotípica5-7 puede ser la causa que explique la escasa frecuencia de anomalías oftalmológicas en estos pacientes, en particular esta paciente solo tiene miopía que se ha mantenido estable. No obstante, las alteraciones oculares tienen una fuerte variación individual.8
El síndrome de Cohen tiene una amplia variedad en sus manifestaciones, por eso tiene gran utilidad realizar un diagnóstico diferencial oportuno, incluso en etapas precoces de la vida, con todos aquellos síndromes que cursan con obesidad, discapacidad intelectual de grado variable, talla baja, hipogenitalismo e hipotonía.
Es de gran importancia realizar un diagnóstico precoz, para poder realizar un asesoramiento genético a las familias y con esto poder actuar más temprano para su mejor incorporación escolar y social.
