INTRODUCCIÓN
El patronímico de atresia pulmonar con septo interventricular intacto (APSI) sugiere una sencilla malformación congénita del corazón en la que todo puede o debe funcionar simplemente al abrir el tracto pulmonar atrésico. Nada más alejado de la realidad que esta suposición, ya que la entidad nosológica se encuentra caracterizada por una gran heterogeneidad en términos de morfología del ventrículo derecho, válvula tricúspide, endomiocardio e, incluso, por una peculiar circulación coronaria.1
Hunter (1884) efectuó el primer reporte al describir un corazón con emergencia pulmonar atrésica y cavidad ventricular derecha sumamente hipertrófica e hipoplásica. El primer intento clasificatorio de la entidad que, curiosamente, conserva plena vigencia, correspondió a Greenwold (1956) quien lo hizo público en su tesis de grado.
Desde los puntos de vista clínico y epidemiológico, la APSI resulta considerada como una cardiopatía congénita crítica con circulación sanguínea pulmonar dependiente del conducto arterioso; representa 1 % del total de las cardiopatías congénitas que se observan en la vida extrauterina y exhibe una distribución acorde a género cercana a 1.5:1 favorable al sexo masculino. Su prevalencia oscila entre 4,2 y 8,5 x 100 000 nacidos vivos; no obstante, a ella corresponde el 3 % del total de cardiopatías congénitas severas, que tienen expresión clínica al nacimiento; además, ocupa el tercer lugar dentro de las malformaciones cardiovasculares cianóticas más frecuentemente detectadas en el período neonatal, solo superada por la transposición de grandes arterias y la atresia pulmonar con septo interventricular abierto.1 El amplio diapasón morfológico de esta entidad torna complejo el análisis al momento de definir la conducta más adecuada a tomar en consideración ante cada caso.
Esta es la causa fundamental de que, a pesar de los avances científicos y técnicos de nuestros días, no se hayan reportado resultados halagüeños en su acometimiento terapéutico, y la dolencia constituya un verdadero reto para la medicina contemporánea, sobre todo para la cardiología y la cardiocirugía pediátricas en cualquier latitud.
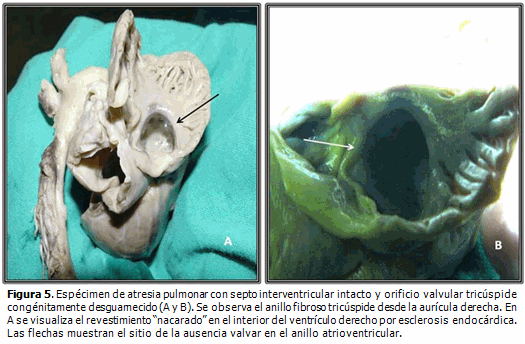
Dentro de las peculiaridades morfológicas de esta enfermedad, resalta la presencia de anillo tricúspide frecuentemente hipoplásico, que correlaciona con el nivel de hipodesarrollo del ventrículo derecho. La válvula tricúspide puede exhibir displasia, presentar valvas prolapsadas o exhibir configuración tipo Ebstein. Raramente se observa su ausencia congénita.2 La existencia de orificio valvular tricúspide congénitamente desguarnecido, forma parte integrante del grupo de malformaciones cardíacas congénitas menos detectadas en la práctica médica cotidiana. Se caracteriza por la ausencia de tejido valvar en el sitio de la normal unión atrioventricular y su correspondiente anillo fibroso (Figuras 1, 2 y 3B). Desde el punto de vista hemodinámico tal situación origina grave regurgitación tricúspide con dilatación auricular derecha consecutiva2,3 (Figura 4). Klein (1938) realizó el primer reporte de la entidad, al observar en la necropsia de una niña de cuatro años de edad, la presencia de anillo tricúspide desprovisto de valvas en conjunción a APSI.2–5
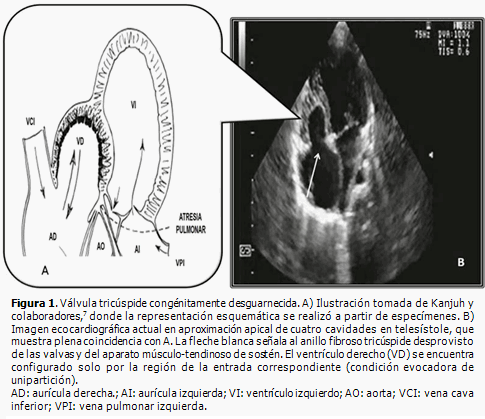
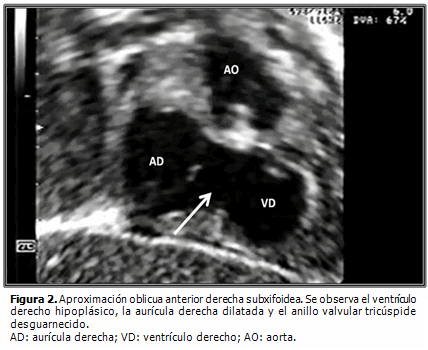
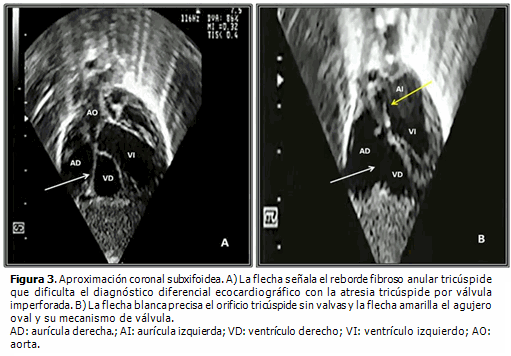
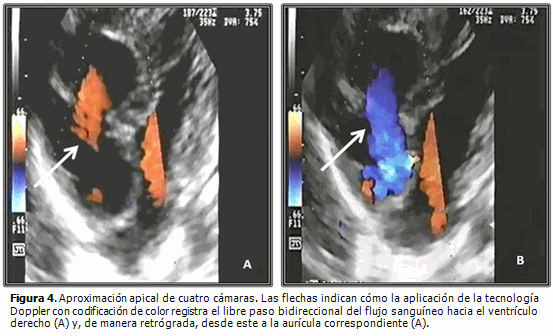
Resulta obvia la importancia que la detección temprana de este hallazgo reviste a la hora de trazar la estrategia terapéutica inicial a implementar (quirúrgica, intervencionista o híbrida), ante cada caso con APSI y al momento de definir las posibilidades reconstructivas o solo paliativas a largo plazo.1,4,6
La presente comunicación se propone como objetivo describir la serie de cuatro pacientes afectados por esta entidad (dos de ellos diagnosticados mediante ecocardiografía), en el Cardiocentro Pediátrico William Soler de la Habana, Cuba, en el período comprendido de 1986 a 2012.
PRESENTACIÓN DE LOS CASOS
De noviembre de 1986 a diciembre de 2012, cuatro pacientes (un niño y tres niñas) con orificio valvular tricúspide congénitamente desguarnecido asociado a atresia pulmonar fueron atendidos en el Cardiocentro Pediátrico William Soler. La edad en días de nacido (mediana 5,00; rango intercuartílico 3,00 – 36,50) y el peso en kilogramos (mediana 3,10; rango intercuartílico 2,90 – 3,75), resultaron compatibles con la presencia de cardiopatía congénita crítica de expresión clínica en época temprana de la vida, dada por cianosis generalizada grave desde el periodo neonatal precoz en todos los pacientes; dos de ellos manifestaron signos de insuficiencia cardíaca derecha debido a restricción del agujero oval.
Tres niños mostraron asociación con la APSI y en el restante se detectó la existencia concomitante de atresia pulmonar con defecto septal interventricular muy pequeño. El diagnóstico ecocardiográfico pudo ser efectuado con precisión en los últimos dos pacientes; la determinación en los dos casos iniciales correspondió a los hallazgos obtenidos de la necropsia.
A todos los pacientes se les practicó fístula quirúrgica de Blalock – Taussig como tratamiento paliativo inicial, en dos de ellos asociada a atrioseptostomía interauricular de balón (por manifestaciones restrictivas del agujero oval). Solo uno de estos últimos niños sobrevivió; las causas de muerte reportadas fueron infarto cerebral sumado a hemorragia pulmonar segmentaria, fibrilación ventricular e insuficiencia respiratoria refractaria al tratamiento respectivamente.
Los resultados de la necropsia mostraron, invariablemente, orificio tricúspide anular de adecuado tamaño con ausencia total de tejido valvar y del correspondiente aparato valvular intraventricular de sostén, marcada hipoplasia ventricular derecha con pérdida de la normal trabeculación y esclerosis endocárdica, atresia pulmonar (valvular o infundibular), dilatación de aurícula derecha e hipertrofia de ventrículo izquierdo (Figura 5).
DISCUSIÓN
Kanjuh y colaboradores (1964) propusieron, para la anomalía, el nombre “orificio valvular tricúspide congénitamente desguarnecido”, apelativo que define la anatomía de la malformación ante la confusión ocasionada por el término “agenesia de la válvula tricúspide”, el cual involucra al trastorno morfológico en cuestión y a la variante de atresia tricúspide ocasionada por ausencia de conexión atrioventricular.7
El desarrollo del aparato valvar tricúspide comienza en la quinta. semana de vida embrionaria con el inicio del proceso de apoptosis local y excavación del tejido miocárdico que rodea el anillo fibroso; la sistemática cavitación parietal del ventrículo derecho, origina redes de haces musculares que se diferencian, de forma sucesiva, en trabéculas carnosas, músculos papilares, cuerdas tendinosas y valvas. Al término de la octava semana de vida intrauterina, se completa la estructura valvular atrioventricular.1,2,4,5 Castellanos y colaboradores señalan, como causa de esta anomalía, la probable incidencia de eventos patogenéticos que provocan la detención inicial de la morfogénesis descrita.8
La mayoría de las comunicaciones efectuadas hasta la fecha, se limitan a la documentación de los hallazgos de la necropsia; la literatura en lengua inglesa registra menos de veinte reportes en vida de los pacientes, que incluyen fetos y, sobre todo, neonatos.9 Anderson y colaboradores (1990) publicaron una serie de 46 casos de APSI, que incluyó tress pacientes con orificio valvular tricúspide congénitamente desguarnecido.3 En veintiséis años de labor, el Cardiocentro Pediátrico William Soler ha recibido, diagnosticado y tratado 67 niños con APSI, de los cuales también tres presentaron la anomalía tricúspide en estudio. Dada la asociación frecuente con APSI y las conocidas limitaciones anatómicas del ventrículo derecho, diversos reportes señalan que estos pacientes han sido sometidos a variantes terapéuticas paliativas, entre las que se encuentran la aplicación de fístula sistémico-pulmonar o la implantación de dispositivos de liberación controlada (stent) en el conducto arterioso, con vistas a garantizar el flujo sanguíneo hacia la circulación arterial pulmonar y la preparación para soluciones quirúrgicas univentriculares a mediano plazo.1,4,6,9
La letalidad tras esos procedimientos resulta aún elevada.
El diagnóstico diferencial ecocardiográfico de esta malformación, puede ser engorroso; incluye el análisis comparativo con la anomalía de Ebstein, la atresia tricúspide por válvula atrioventricular derecha imperforada (Figura 4A), la enfermedad de Uhl y la displasia arritmogénica del ventrículo derecho.2–4,6,10
En la anomalía de Ebstein:
- Se aprecian inserciones de las valvas septal y mural en el ventrículo derecho, más que en la unión atrioventricular; esto se debe a fallas embriológicas de liberación de estas valvas desde la pared ventricular.
- Se puede observar el diferente nivel de implantación valvar septal (bajo) respecto al anterosuperior (normal).
En la atresia tricúspide por válvula imperforada:
- Se visualiza la membrana o diafragma que ocluye el orificio tricúspide. Esta estructura no debe ser confundida con el reborde anular atrioventricular adyacente (Figura 3A).
En la enfermedad de Uhl:
- Existe ausencia de miocardio parietal en el ventrículo derecho con preservación de las trabeculaciones septales y normalidad estructural de las válvulas tricúspide y sigmoidea pulmonar.
En la displasia arritmogénica de ventrículo derecho:
- Presencia de degeneración miocárdica de ventrículo derecho con reemplazo por tejido fibroadiposo.
- La válvula tricúspide es normal.
La serie de pacientes presentados cumple los criterios planteados con anterioridad en esta discusión. Se insiste en recalcar la importancia que reviste la identificación ecocardiográfica temprana de esta malformación asociada a la APSI para el pronóstico vital de los niños que padecen esta última entidad mencionada.
Con vistas a garantizar el pronóstico vital de los niños que padecen de APSI, se insiste en subrayar la importancia que reviste la identificación ecocardiográfica temprana de ausencia de desarrollo valvular tricúspide asociada.
