INTRODUCCIÓN
Dentro de las numerosas aplicaciones médicas de la citogenética, el diagnóstico prenatal citogenético (DPC) constituye tal vez el punto más crítico, debido a que de sus resultados se deriva una importante decisión para la familia: continuar o no un embarazo.1
Al encontrar en una misma muestra de líquido amniótico células con cariotipo normal y células con cariotipo anormal, surge la sospecha de un mosaicismo cromosómico. En consecuencia, debe hacerse una correcta distinción entre un mosaico cromosómico verdadero que representa una anomalía in vivo y un pseudomosaicismo debido a errores en la mitosis de las células del cultivo (in vitro), u otros factores, como las células que se derivan del tejido extraembrionario (placenta, saco vitelino, cordón umbilical, etc).1
Entre las posibles anomalías cromosómicas detectables por el DPC se encuentra el mosaicismo cromosómico, que puede definirse como la presencia en un individuo o tejido de al menos dos líneas celulares que difieren en su cariotipo, derivando ambas de un cigoto único.1
En estos casos la estrategia a seguir debe tomar un carácter multidisciplinar, interviniendo citogenetistas, especialistas en Biología Molecular, genetistas clínicos, obstetras y especialistas en ecografía.1
Durante la realización del DPC se siembran al menos tres frascos de cultivo por cada paciente remitida al laboratorio. Si la línea celular aberrante aparece en al menos dos frascos de cultivo diferentes se puede definir como un mosaico verdadero.1
El Síndrome de Turner tiene una prevalencia de 1 en 1800 a 5000 recién nacidos vivos femeninos y se caracteriza por la ausencia total o parcial del segundo cromosoma X. Actualmente se reconoce una gran variedad en su presentación citogenética, siendo la más común la monosomía del X (45, X); entre las menos frecuentes están los mosaicismos que incluyen fragmentos o la totalidad del cromosoma Y. La presencia de este cromosoma confiere características fenotípicas de androgenización, por lo que se requieren estudios adicionales en estos pacientes.2
Es aconsejable la realización de un ultrasonido prenatal detallado de los genitales. De no encontrarse alteraciones, es alta la probabilidad de un fenotipo de varón normal.1
El desarrollo hacia ovarios o testículos está determinado por la acción coordinada de una secuencia de genes que conllevan a la diferenciación gonadal femenina cuando no existe el cromosoma Y, o el desarrollo testicular cuando este cromosoma está presente. En 1987 Affara y colaboradores probaron con sondas complementarias a la región del brazo corto de este cromosoma en individuos fenotípicamente varones, pero que presentaban cromosomas sexuales XX, y descubrieron que dichas sondas hibridizan, es decir, que eran complementarias al ADN de estos individuos.3 Este resultado los llevó a deducir la presencia de material genético del brazo corto del cromosoma Y en estas personas, logrando establecer una región responsable del desarrollo testicular en el brazo corto del cromosoma Y, denominada Yp11.3.3 Más tarde, Page determinó que en esta región se ubica un gen al que se llamó SRY (por las siglas en inglés de sex-determining region on the chromosome Y), el cual codifica para un factor de transcripción que actúa a modo de interruptor e inicia la cascada de desarrollo gonadal masculino.4,5-7
Cercana a el gen SRY se encuentra una región pseudoautosómica en la que se realiza intercambio de material genético durante la meiosis, entre el cromosoma Y y el cromosoma X en el espermatogonio, proceso que se conoce como entrecruzamiento. Page y colaboradores, en 1989, así como otros autores 4 logran determinar que al darse el entrecruzamiento entre los cromosomas X y Y, algunas veces ocurre intercambio del gen SRY entre estos cromosomas, ocurriendo pérdida del gen por parte del cromosoma Y, así como ganancia de este por parte del cromosoma X, explicándose el por qué individuos XX lograban desarrollar testículos y mujeres fenotípicas con cariotipos XY no los desarrollaban.4,7,8
En este artículo se expone el resultado de un diagnóstico prenatal de esta aberración cromosómica.
PRESENTACIÓN DEL CASO
En el mes de enero del año 2011 se realizó amniocentesis para DPC (DPC) en el Centro Provincial de Genética Médica de Cienfuegos (CPGMC) a una gestante de 42 años de edad. El resultado del DPC fue 45,X/46,XX. Las láminas extendidas del DPC de esta embarazada fueron tomadas como control de calidad del laboratorio y enviadas al Centro Nacional de Genética Médica (CNGM), donde se corroboraría el resultado. (Figs. 1 y 2)
En ultrasonidos realizados a la embarazada siempre aparentaba feto de sexo masculino, por lo que se decidió repetir la amniocentesis para realizar diagnóstico molecular de sexo en el CNGM mediante la técnica de Reacción en Cadena de la Polimerasa (PCR)5, con vistas a comprobar si el feto era portador de este gen, quedando como diagnóstico final mos 45,X[40]/46,XX[10] (mosaico cromosómico en el que se vieron al microscopio 40 células 45,X, 10 células 46,XX) con presencia del gen SRY.
Luego de informar del diagnóstico a la pareja, se le brindó asesoramiento genético. Decidieron interrumpir el embarazo a las 25, 3 semanas de gestación.
El estudio anátomo patológico informó sobre cadáver de feto de 820 gramos de peso y 25,3 semanas de gestación, que presentó armonía corporal generalizada, pene morfológicamente normal con prepucio discreto, glande y meato normales. Bulbos escrotales normales que a la palpación estaban vacíos. En la zona genital interna se identificó una masa oval tabicada ubicada por debajo de la vejiga que corresponde a próstata hipoplásica acompañada de las vesículas seminales ligeramente dilatadas y ausencia testicular. No pudo realizarse el estudio cromosómico en el material abortivo por no tener las condiciones necesarias para esto, por lo que no fue posible comprobar el diagnóstico citogenético postmorten.
La paciente fue informada acerca del interés científico para el CPGM del reporte de su caso y dio su consentimiento para la confección y divulgación del informe (Anexo 1).
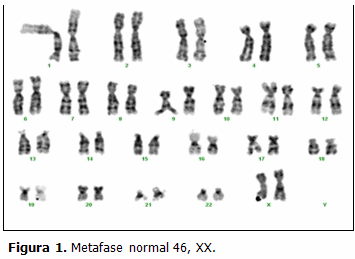
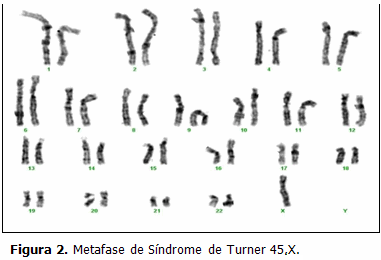
DISCUSIÓN
No existe una correlación directa entre la proporción de células con cariotipo 45, X y el fenotipo en los casos de mosaicismo 45, X/46, XY, ejemplo de ello es un caso reportado en Cuba por el CNGM, aunque en el estudio del material abortivo solo se observaron células masculinas;1 y en otro caso reportado en Colombia de una paciente adulta que acudió a la consulta por amenorrea primaria, genitales externos femeninos de apariencia usual, pero que al realizarle una ecografía no se encontró útero ni ovarios.2 Ambos ejemplos presentaban mosaicismo 45, X/46, XY y sin embargo fenotípicamente eran de sexos diferentes.
Anteriormente se han reportado estudios en pacientes con ST, tanto en su forma clásica 45, X, como en mosaico 45X/46XX, buscando la posible presencia del gen determinante del sexo, pero solo se ha hallado en muy pocos casos,9 aún cuando la técnica del PCR tiene una sensibilidad capaz de detectar una célula con contenido de cromosoma Y entre un 1 000 000 de células.10
Por lo ya expuesto, nuestro caso puede explicarse por dos hipótesis: Puede ser un mosaicismo críptico que aparece en muy baja proporción, donde solo se afecta un número limitado de tejidos en la persona;1,9,11 o puede existir material genético del brazo corto del cromosoma Y en uno de los cromosomas X como consecuencia de un error ocurrido en el entrecruzamiento de las regiones seudoautosómicas de los cromosomas X y Y, donde la totalidad o gran parte del gen SRY es transferida al cromosoma X, lo cual pudiera explicar el cariotipo 45,X/ 46, XX con presencia del gen SRY presentado en este caso.4,7
Es recomendable indicar el diagnóstico molecular de sexo en este tipo de caso, con la finalidad de buscar la presencia de anomalías en la diferenciación sexual.12 En el caso expuesto, la presencia de pene, más una masa quística que pudiera corresponder a una próstata hipoplásica y a la vesícula seminal, junto con estructuras internas y externas masculinas, y ausencia de testículos, sumado a los resultados arrojados por los complementarios, evidencian la existencia de una anomalía en la diferenciación sexual.

