INTRODUCCIÓN
En situaciones fisiológicas la sangre se mantiene en estado líquido dentro de la vasculatura y al mismo tiempo es capaz de formar coágulos para sellar una herida. (1)
El término hemostasia significa prevención de la pérdida de sangre. Siempre que se lesiona un vaso, la hemostasia se consigue por diversos mecanismos: espasmo vascular, formación de un tapón de plaquetas, formación de un coágulo sanguíneo y proliferación final de tejido fibroso para el completo cierre de la lesión.
La coagulación de la sangre es un proceso delicadamente equilibrado que resulta de una serie ordenada de reacciones, en el cual existe participación e interacción entre células y proteínas con características bioquímicas especiales. (1)
El modelo clásico de la coagulación fue descrito en 1964 por Davie y Ratnoff como dos secuencias de reacciones lineales e independientes entre sí, que culminaban en una vía final común con la activación del factor X. De acuerdo con este modelo, la activación de cualquiera de las dos vías resultaba en la producción de grandes cantidades de trombina y la subsecuente formación de fibrina. (2)
El modelo fue muy útil al describir de forma organizada la interacción entre las proteínas con actividad pro-coagulante y probablemente siga encontrando utilidad al apoyar la evaluación de los tiempos globales de la coagulación. (2)
Sin embargo, este modelo no es válido para explicar los mecanismos que llevan a la hemostasia in vivo; no le otorga importancia a cada uno de los complejos con actividad pro-coagulante; no considera la interacción del sistema con las células que participan en la coagulación; no considera las interacciones entre las dos vías de la coagulación y falla en explicar con detalle los aspectos fisiopatológicos del sistema hemostático. En otras palabras, el modelo no permite explicar los distintos grados de tendencia a la hemorragia que resultan de deficiencias de los diferentes componentes de las dos vías. (3)
¿Por qué la deficiencia de factor XII no produce problemas de sangrado?, ¿por qué en la hemofilia el factor VII endógeno no compensa la falta de los factores deficientes para la producción de trombina? Son sólo dos de las muchas cuestiones que el modelo tradicional no puede contestar.
En un intento por abordar el fenómeno de la hemostasia desde otra perspectiva, se han desarrollado modelos experimentales y conceptuales para probar las hipótesis en un modelo bioquímico ex vivo, y permitir un mejor entendimiento de cómo el sistema funciona in vivo.
El más logrado de éstos es el modelo celular de la coagulación desarrollado por Maureane Hoffman en el 2003 donde el aspecto más importante es considerar a las células como elementos esenciales en el proceso de formación del coágulo y demostrar que las superficies celulares poseen características especiales capaces de dirigir el proceso hemostático. La nueva teoría rompe así con el paradigma del modelo tradicional de Davie y Ratnoff en el cual el papel de la célula era únicamente el de ofrecer una superficie portadora de fosfatidilserina donde los complejos pro-coagulantes podrían ser armados. (4)
Todo lo anterior justifica y sustenta la importancia de esta revisión bibliográfica, debido a que la teoría tradicional de las cascadas de la coagulación conocida y empleada por decenas de años está siendo sustituida por un nuevo y complejo modelo celular que responde a todas las interrogantes que existían en relación con este tema.
Por todo lo anterior queda implícito el fundamento de esta revisión y la elección de esta temática, ya que representa una nueva ventana de exploración para poder describir y comprender efectivamente el mecanismo de la coagulación, proceso esencial para mantener la adecuada hemostasia del organismo. Por ello se pretende describir los aspectos más importantes de la nueva teoría celular de la coagulación y sus ventajas respecto a la vieja teoría, destacar los principales elementos de la teoría clásica de la coagulación así como las diferentes fases que conforman este nuevo modelo integrador de la hemostasia. Se comenta además sobre las perspectivas clínicas del modelo celular de la coagulación.
DESARROLLO
ASPECTOS IMPORTANTES DE LA COAGULACIÓN
La coagulación consiste en una serie de reacciones que se generan en la superficie celular y cuyo objetivo es la formación de trombina en sitios de lesión vascular. Es un proceso delicadamente equilibrado en el cual existe participación e interacciones entre células y proteínas con características bioquímicas especiales (también conocidas como factores de la coagulación), resaltando la importancia del complejo factor VII/factor tisular en la activación del sistema. (1)
Los factores de la coagulación dependientes de vitamina K, comparten características bioquímicas y estructurales especiales; la más importante de estas es la presencia de un dominio de ácido -carboxiglutámico en la región amino-terminal de la molécula. Este dominio contiene entre 8 y 12 residuos de glutamato (Gla) y tiene 3 funciones de gran importancia fisiológica: 1) permitir la activación de la proteína a través de la carboxilación de residuos de ácido glutámico; 2) favorecer la unión con iones de calcio y otros cofactores para catalizar las reacciones de proteólisis; 3) facilitar la interacción de los fosfolípidos de carga negativa para aumentar la actividad proteolítica. (1)
Además de la estructura, estos factores de la coagulación comparten características funcionales especiales; todos son sintetizados en el hígado y sufren cambios postranscripcionales consistentes en: eliminación del propéptido señal y la mencionada carboxilación de los residuos de ácido glutámico a través de la enzima glutamato-carboxilasa.
Estos factores circulan en forma de cimógenos o proenzimas que al activarse adquieren capacidad de proteasa de serina, la cual se ve potencializada por la presencia de cofactores específicos. (5)
De la misma manera, la asociación de estas enzimas con las cabezas con carga negativa de los fosfolípidos de membrana, especialmente la fosfatidilserina, incrementa la actividad de proteasa. (5)
FACTORES DE LA COAGULACIÓN
Factor II: la trombina es la enzima efectora central del sistema de coagulación al tener varias funciones importantes: a) la función principal y más conocida de la trombina es la escisión de los fibrinopéptidos A y B, los cuales se polimerizan para formar la fibrina; b) es un potente activador de plaquetas a través de receptores PAR-1 y PAR-4, así como de la glucoproteína Ib; c) tiene efectos procoagulantes al participar en la retroalimentación positiva mediante la activación de los factores V, VIII, XI y XIII; d) activa a la enzima parecida a procarboxipeptidasa-B, también conocida como inhibidor de fibrinólisis activado por trombina (IFAT), la cual inhibe la degradación de fibrina mediada por plasmina; e) agregado a sus efectos pro-coagulantes la trombina se une a su cofactor celular, trombomodulina, presente en las células endoteliales de los lechos microvasculares, lo que permite la activación de la proteína; f) son también conocidas las actividades de factor de crecimiento y de citocina con un papel creciente en los procesos de aterosclerosis, reparación de heridas e inflamación. La protrombina es escindida por el complejo protrombinasa, que consiste en un complejo unido a fosfolípidos formado por la enzima factor Xa y su cofactor Va. El dominio efector (trombina) se separa del resto de la proteína. El principal inhibidor plasmático de la trombina es la antitrobina III. (5)
Factor VII/factor tisular: conocido como proconvertina, al factor VII actualmente se le considera la piedra angular de la activación de los procesos de hemostasia, junto con su cofactor, el factor tisular. La mayor parte del factor VII se encuentra en la sangre en forma de cimógeno y sólo un 1 % circula de manera activa. Su principal activador es el factor X.
El factor tisular es una proteína de membrana presente de manera abundante en las células que rodean el lecho vascular, sobre todo fibroblastos y músculo liso; es el único factor de la coagulación que normalmente no está presente en la sangre. Aunque algunos estudios señalan su presencia en las membranas de leucocitos y monocitos.
El factor VIIa y el factor tisular se ponen en contacto cuando existe lesión vascular, el complejo activa los factores IX y X y es inhibido principalmente por la vía del inhibidor del factor tisular (VIFT) y en menor medida por la antitrombina III. (6)
Factor IX/factor VIII: el factor IX es una enzima fundamental en los procesos de hemostasia y su ausencia congénita se traduce clínicamente en tendencia al sangrado (hemofilia B, deficiencia de Christmas). Tiene dos fuentes potenciales de activación: el complejo factor VIIa/FT y el factor XIa, existe también una glicoproteína plaquetaria con la capacidad de activar este factor.
Pequeñas cantidades de factor IX son activadas de forma basal por el complejo VIIa/FT fisiológicamente, pero no está claro el papel potencial del factor IXa en los procesos de activación de la coagulación. El factor VIII es una proteína con actividad de cofactor soluble, que viaja unido al factor de von Willebrand, lo que le confiere una mayor vida media.
Es activado por la trombina y por el factor Xa. Una vez activados, el factor IX se une con el factor VIII, que junto con Ca+ y en presencia de fosfolípidos constituyen el complejo Xasa. (5, 6)
Factor X/factor V: el factor de Stuart-Prower, como se le conocía anteriormente, es una proteasa de serina que, junto con el cofactor Va y fosfolípidos de membrana, forma el complejo protrombinasas que activa a la trombina. Representa el primer factor de la vía final común en el modelo antiguo de la hemostasia y tiene, de la misma manera, dos fuentes potenciales de activación: el complejo factor VIIa/FT y el complejo IXa/VIIIa.
El factor V es homólogo al factor VIII en su estructura génica, secuencia de aminoácidos y dominios moleculares. Circula en forma libre en el plasma, pero un 20 % se encuentra en los gránulos plaquetarios. Su principal activador es la trombina, pero puede también ser activado por el factor X. (7)
Fibrinógeno y factor XIII: el fibrinógeno es una glucoproteína perteneciente al grupo de las globulinas, presente en el plasma en grandes concentraciones (300-400 mg/dl), y en menor medida en los gránulos alfa de las plaquetas. Su síntesis corre a cargo del hepatocito y está influenciada por estímulos inflamatorios. Al ser escindido por la trombina, libera los fibrinopéptidos A y B, que forman la fibrina, las que al polimerizarse de forma espontánea forman una red que cubre y da resistencia al coágulo. (8)
El factor XIII es igualmente una glucoproteína formada por dos subunidades y cuya función es entrecruzar las cadenas y las de la fibrina para estabilizar el coágulo y protegerlo contra las acciones de la plasmina. (9)
ARN CON PAPEL SINGULAR EN LA HEMOSTASIA
Las proteínas son las moléculas biológicas por excelencia, ya que son los constituyentes característicos de las estructuras celulares y son también las especies que llevan a cabo las funciones celulares más importantes. Se encuentran codificadas en el ADN, pero desde el código genético hasta la correcta ejecución de una función celular realizada por una proteína median muchos pasos que son objeto de una estrecha regulación. (10)
El ARNm es el molde utilizado para la síntesis de proteínas, trasladando la información genética desde el ADN del núcleo a los ribosomas, donde se sintetizan las cadenas polipeptídicas que dan lugar a las proteínas.
En la última década se ha caracterizado una familia de ARN no codificantes relacionada con gran cantidad de procesos biológicos que, por su pequeño tamaño, ha recibido el nombre de microARN (miARN). Un miARN está formado por una cadena sencilla de ARN de unos 19-22 nucleótidos, capaz de controlar la expresión génica fundamentalmente a nivel postranscripcional (degradando el ARNm o inhibiendo su traducción, según el grado de complementariedad). (11)
Aunque resulta atractiva la hipótesis de que la amplia variabilidad observada en los niveles circulantes de los factores hemostáticos se encuentre regulada por miARN, hasta la fecha actual no hay evidencias que la sustenten. Por el contrario, sí existen trabajos publicados que describen el patrón de expresión de miARN en las diferentes líneas celulares sanguíneas tanto en el proceso de diferenciación, como en líneas maduras circulantes y en situaciones patológicas como linfomas, leucemias y la policitemia vera, entre otras. En particular en el proceso de hemostasia primaria se han publicado datos preliminares que apoyan la participación de los miARN en la activación plaquetaria. Así, aunque las plaquetas no poseen ADN genómico retienen cierta cantidad de ARNm procedente de los megacariocitos que puede ser sometida al control de los miARN. (12)
Recientemente se ha descrito que el daño tisular puede también liberar ARN a partir de las células lesionadas, que al alcanzar el torrente circulatorio puede exhibir una actividad pro-coagulante. (13)
SERPINAS HEMOSTÁTICAS
La hemostasia y la fibrinólisis son los sistemas que garantizan un correcto flujo sanguíneo, así como una adecuada respuesta pro-coagulante cuando es necesaria, mediante una red de procesos fisiológicos interconectados y una sucesión de reacciones proteolíticas. Las reacciones enzimáticas que promueven estas rutas son catalizadas por serinaproteasas, que necesariamente deben ser controladas por diferentes tipos de inhibidores. (14)
Gran parte de este control, trascendental para una correcta hemostasia, se realiza por un grupo de inhibidores conocidos como serpinas (acrónimo de las palabras “serine protease inhibitors”) y sus cofactores. (15)
Las serpinas son una superfamilia de proteínas clasificadas dentro de 16 secciones (A-P). El genoma humano contiene aproximadamente 36 serpinas implicadas en la regulación de numerosos sistemas que incluyen la hemostasia y fibrinólisis, angiogénesis, la cascada del complemento y la inflamación. (15)
Todas ellas presentan un alto grado de homología estructural. Están típicamente compuestas por aproximadamente 400 aminoácidos que se organizan en 9 hélices (A-I) y tres hojas β (A-C).
Un esquema minimalista de la cinética de inhibición de las serpinas consta de dos pasos: 1) la formación de un complejo michaeliano, donde la secuencia del loop reactivo (RCL) es reconocida por la proteasa como un sustrato; y 2) la formación de un complejo covalente donde la proteasa es atrapada en un estado inactivo. (16)
Puesto que la especificidad de la serpina se determina ampliamente por la frecuencia de formación del complejo michaeliano, los cofactores que se unen a las serpinas (y a veces a la proteasa objeto) pueden alterar radicalmente la especificidad. El cofactor mejor entendido para las serpinas es la heparina y sustenta el potencial terapéutico anticoagulante de esta. La heparina se une a la mayoría de las serpinas implicadas en hemostasia y trombosis. (16)
Las serpinas con papel importante en la hemostasia son:
- 1-antitripsina (1 AT): Serpina 1. Su diana fisiológica es la elastasa de neutrófilos, pero también inhibe a la proteína C activada (APC) de forma independiente de heparina. Pero en términos generales no se da mucho valor a la contribución de la 1-AT en la coagulación. Destacamos la variante Pittsburgh que es un potente inhibidor de diversas serinaproteasas de la cascada de la coagulación, especialmente de la trombina y de la APC. (16)
- Antitrombina: Serpinc 1. Se trata sin duda de la principal serpina hemostática y el inhibidor fisiológico más importante de la cascada de la coagulación. Como su nombre indica es el principal inhibidor de trombina, pero también inhibe FVIIa, FIXa, FXa, FXIa y FXIIa. Su actividad anticoagulante predominante se debe a la inhibición de FXa y trombina. (17)
- Cofactor II de heparina (CIIH): Serpind 1. Inhibe a la trombina en presencia de diferentes moléculas polianiónicas incluyendo a la heparina, heparán sulfato y dermatán sulfato. De hecho, el 20-30 % de la inhibición de la trombina en la coagulación es mediada por el CIIH. (18)
- Inhibidor de la proteasa dependiente de proteína Z (ZPI): Serpina 10. El ZPI es una glicoproteína hepática de 72 kDa, que se secreta al plasma, donde alcanza una concentración de 1,5 µg/mL. En presencia de proteína Z, fosfolípidos y calcio, el ZPI inhibe rápidamente al FXa. En ausencia de cofactores, el ZPI inhibe además al FXIa y su inhibición puede acelerarse dos veces mediante su unión a heparina.
- Inhibidor de la proteina C (PCI): Serpina 5. El PCI es una proteína de unión a heparina que inhibe múltiples proteasas, incluyendo el APC, trombina libre y trombina unida a trombomodulina, por tanto, desempeña tanto funciones anticoagulantes como pro coagulantes, dependiendo de la proteasa diana y de la presencia de cofactores específicos.
- 2-antiplasmina (2-AP): Serpinf 2. Es el principal inhibidor fisiológico de la plasmina.
- Inhibidor del activador de plasminógeno-1 (PAI-1): Serpine 1. Es la principal serpina fibrinolítica sintetizada en las células endoteliales, plaquetas y otras células mesenquimales que rodean la vasculatura.
TEORÍA CLÁSICA DE LA COAGULACIÓN
La coagulación de la sangre se produce en tres pasos fundamentales:
- En respuesta a la ruptura o a la lesión de un vaso sanguíneo se forman unas sustancias, que constituyen el llamado complejo activador de la protrombina.
- El activador de la protrombina cataliza la transformación de la protrombina en trombina.
- La trombina actúa como una enzima para convertir el fibrinógeno en fibras de fibrina, que atrapan plaquetas, eritrocitos y plasma para formar el coágulo.
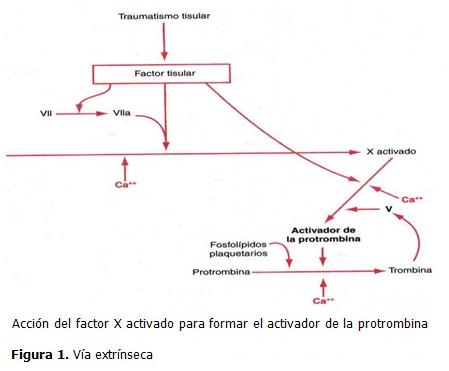
Al iniciarse la coagulación se forma el activador de la protrombina el cual puede producirse por dos vías: 1) la vía extrínseca, que comienza con un traumatismo de la pared vascular y de los tejidos circundantes y 2) la vía intrínseca, que se inicia en la propia sangre. (2, 19)
El mecanismo extrínseco para el inicio de la formación del activador de la protrombina comienza cuando la pared vascular o un tejido extravascular sufren un traumatismo y se produce mediante los tres pasos siguientes: (20, 21)
- Liberación de tromboplastina tisular. El tejido lesionado libera un complejo de varios factores, llamado tromboplastina tisular; estos factores son fosfolípidos de las membranas de los tejidos dañados y un complejo lipoproteico que actúa como enzima proteolítica.
- Activación del factor X para formar factor X activado. El complejo lipoproteico de la tromboplastina tisular se combina con el factor VII de la coagulación y en presencia de los fosfolípidos de los tejidos dañados y de iones calcio, actúa enzimáticamente sobre el factor X para dar factor X activado.
- Efecto del factor X activado para formar el activador de la protrombina. El factor X activado se combina inmediatamente con los fosfolípidos tisulares liberados, que forman parte de la tromboplastina tisular y con el factor V para formar el complejo llamado activador de la protrombina. A los pocos segundos, este escinde la protrombina para formar trombina y el proceso de coagulación prosigue como se ha descrito. El factor X activado es la proteasa que realmente produce la ruptura de la protrombina para dar trombina. (Figura 1)
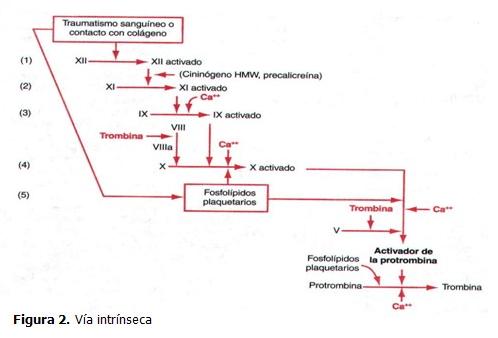
El mecanismo intrínseco para el inicio de la formación del activador de la protrombina comienza con un traumatismo de la propia sangre o con la exposición de la sangre al colágeno de la pared de un vaso sanguíneo lesionado. El proceso se produce mediante la siguiente cascada de reacciones:(20)
- Activación del factor XII y liberación de fosfolípidos plaquetarios. Debido al traumatismo el factor XII se activa para formar una enzima proteolítica llamada factor XII activado. Simultáneamente, el traumatismo sanguíneo daña las plaquetas, por lo que se liberan fosfolípidos plaquetarios que contienen una lipoproteína llamada factor III plaquetario, que interviene en las reacciones de coagulación posteriores.
- Activación del factor XI. El factor XII activado actúa enzimáticamente sobre el factor XI para activarlo. Este segundo paso de la vía intrínseca requiere la presencia de cininógeno de peso molecular elevado (HMW).
- Activación del factor IX por el factor XI activado. El factor XI activado actúa luego enzimáticamente sobre el factor IX para activarlo.
- Activación del factor X. El factor IX activado junto con el factor VIII, los fosfolípidos plaquetarios y el factor III de las plaquetas dañadas, activan al factor X. Este paso de la vía intrínseca es igual que el último de la vía extrínseca, es decir, el factor X activado se combina con el factor V y con los fosfolípidos plaquetarios o tisulares para formar el complejo llamado activador de la protrombina. El activador de la protrombina, a su vez, inicia la escisión de la protrombina para formar trombina, poniendo en marcha el proceso final de la coagulación. (Figura 2)
De lo dicho en párrafos anteriores se desprende un grupo de cuestionamientos básicos sobre la base de los cuales un grupo de investigadores han dado cuerpo al nuevo modelo celular de la coagulación y han tratado de dar explicación a preguntas que estaban aún sin respuesta. Un ejemplo clásico es la poca importancia de la vía intrínseca y al factor XII como su punto clave de inicio, ya que en estudios in vivo en pacientes con deficiencias del XII han primado las manifestaciones trombóticas por encima de las hemorrágicas; se ha podido comprobar también que esta vía y este factor son más bien un enlace de la coagulación con los procesos de activación del complemento y la cascada de la inflamación y no cumplen un rol central en la coagulación. Por otra parte sitúa a la trombina en un papel esencial como una molécula con actividad pleotrópica en el organismo y con un papel central en el proceso de coagulación. Esto explicaría en parte por qué el factor VII endógeno no es suficiente por sí solo para controlar la hemorragia en la hemofilia y aclara de forma más convincente el papel del factor VIII en el proceso de estallido de la producción de trombina, puestos en relieve de forma más clara en la teoría celular de la coagulación.
NUEVO MODELO CELULAR DE LA COAGULACIÓN
En un intento por abordar el fenómeno de la hemostasia desde otra perspectiva, se han desarrollado modelos experimentales y conceptuales para probar las hipótesis en un modelo bioquímico ex vivo, y permitir un mejor entendimiento de cómo el sistema funciona in vivo. El más logrado de estos es el modelo celular de la coagulación desarrollado por Hoffman y cols. El aspecto más importante del modelo es considerar a las células como elementos esenciales en el proceso de formación del coágulo y demostrar que las superficies celulares poseen características especiales capaces de dirigir el proceso hemostático. La nueva teoría rompe así con el paradigma del modelo tradicional, según el cual, el papel de la célula era únicamente el de ofrecer una superficie portadora de fosfatidilserina donde los complejos procoagulantes podrían ser armados. El nuevo modelo, también hace énfasis en que la coagulación ocurre en tres fases, que ocurren simultáneamente en diferentes superficies celulares. (4, 22)
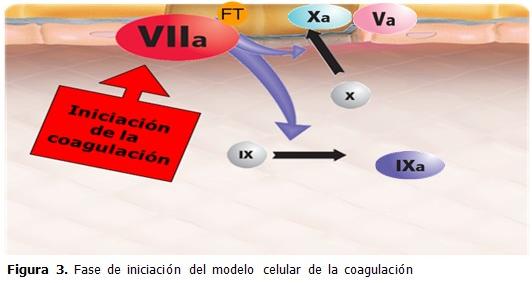
La primera fase de iniciación ocurre en las células portadoras de factor tisular (subendotelial); en la fase de amplificación el sistema se prepara para la producción a gran escala de trombina y finalmente la tercera fase, de propagación, ocurre en la superficie plaquetaria y resulta en la producción de grandes cantidades de trombina.(4,22)
Iniciación
El factor VIIa y el factor tisular son elementos esenciales en el inicio de los procesos de hemostasia. El factor VII circula en la sangre predominantemente como molécula inactiva y sus funciones a las concentraciones fisiológicas, son virtualmente nulas en ausencia de su cofactor. El factor tisular no está en contacto con elementos de la sangre; la célula que alberga este receptor (fibroblasto, miocito, célula mono nuclear, macrófago) se encuentra fuera del sistema vascular hasta que existe pérdida de la integridad del mismo. La interacción entre el factor tisular y el factor VIIa es el proceso fundamental en la iniciación de la coagulación; tal interacción incrementa la actividad del factor VII en 1 x 10(7). El complejo FVIIa/FT activa a los factores X y IX, y el factor Xa formado, es capaz de generar pequeñas cantidades de trombina de manera local.
Existe evidencia que sugiere que estas reacciones responsables de la iniciación de la coagulación ocurren de forma continua fuera de la vasculatura en individuos sanos.
El factor VII, X y la protrombina, son capaces de permear a través de espacios titulares fuera del sistema vascular y pueden ser detectados en linfa y tejidos perivasculares.
Sobre la base de estas observaciones se formuló la teoría de “mínima función”, en la cual el sistema del factor tisular tiene actividad constante, generando constantemente pequeñas cantidades de trombina fuera del sistema vascular en individuos sanos. A pesar de que el paso inicial de la coagulación se produce de manera continua, esto no conduce a la formación de coágulos ya que las reacciones y sus productos se encuentran afuera de la vasculatura y de otros elementos esenciales del sistema; la interacción de unos con otros requiere de una pérdida de integridad de la pared de los vasos. (22) (Figura 3)
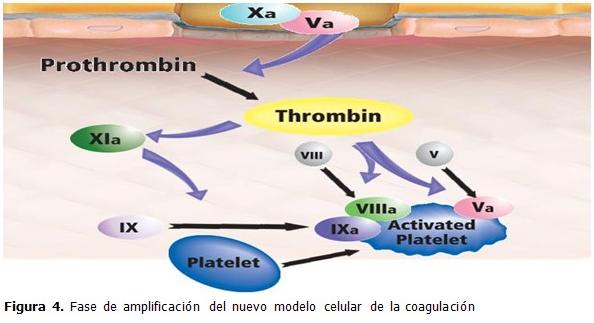
Amplificación
Como resultado de la lesión vascular, los elementos del sistema que son incapaces, por su tamaño, de abandonar el espacio intravascular, están ahora aptos para hacerlo. El más importante de estos es la plaqueta. La fase de amplificación es dependiente de la presencia de membranas plaquetarias activadas y de la interacción de estas con los factores de la coagulación, especialmente con las cantidades limitadas de trombina que se generan en la vecindad de la célula portadora de factor tisular (vide supra). Las plaquetas se activan y degranulan, al tiempo que se adhieren y agregan formando un tapón en el vaso dañado; una característica muy importante en la activación de las plaquetas es el cambio de polaridad de las cabezas negativas de los fosfolípidos para permitir su interacción con los factores de la coagulación.
Aunque es insuficiente para la formación de un coágulo, la pequeña cantidad de trombina producida por la vía VIIa/FT, durante la fase de iniciación, es esencial para amplificar el proceso. La trombina es un ávido reclutador de plaquetas y retroalimenta de manera positiva al sistema al poseer la capacidad de activar a los factores V, VIII y XI. La fase de propagación también se caracteriza por la activación del sistema de retroalimentación negativa a través de los anticoagulantes naturales: VIFT, antitrombina III y proteína C, cuya función es importante en regular los procesos pro-coagulantes. Finalmente el complejo IXa/VIIIa se ensambla en la superficie plaquetaria y genera grandes cantidades de factor X; parte de este complejo se ensambla en la célula portadora de factor tisular y puede difundir a la superficie plaquetaria dada su resistencia relativa a los efectos de anticoagulantes naturales. (22)
El papel de este complejo eventualmente supera la del complejo VIIa/FT en la producción de Xa, ya que es 50 veces más eficiente y dada la inactivación creciente del VIIa/FT por el VIFT. (Figura 4).
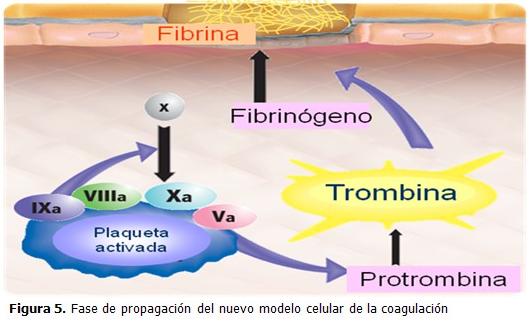
Propagación
La fase de propagación presenta un cambio de locación de los procesos que lleva a la generación de la trombina, de la célula portadora de factor tisular a la plaqueta activada. La presencia de fosfolípidos en la membrana plaquetaria activada permite el ensamblaje del complejo IXa/VIIIa y potencia sus acciones en 1 x 10(8). Grandes cantidades de trombina se producen durante esta fase resultando en la escisión proteolítica del fibrinógeno y formación de monómeros de fibrina que se polimerizan para consolidar el inestable coágulo inicial de plaquetas en un firme coágulo organizado de fibrina. La trombina a su vez activa al factor XIII y al IFAT con efectos positivos adicionales en la estabilidad del coágulo y en la resistencia a los efectos de la plasmina. (22) (Figura 5).
IMPLICACIONES CLÍNICAS DEL NUEVO MODELO
NOVO SEVEN: FACTOR VII RECOMBINANTE ACTIVADO (FVIIRA)
Dado el importante papel que adquiere la actividad del factor VII en el nuevo modelo celular de la coagulación, sobre todo para darle inicio, este factor se está comenzando a emplear en una amplia variedad de enfermedades con compromiso de la hemostasia, con muy buenos resultados.
El factor VII recombinante activado (FVIIra) se desarrolló originalmente para tratar los episodios de sangrado en pacientes hemofílicos. Recientemente reportes de casos han mostrado eficacia en el uso de factor VII para tratar otras causas de hemorragia descontrolada, de etiología diferente. El uso generalizado de este producto ha permitido comprender que el proceso de la coagulación puede ocurrir incluso cuando algunos factores de la coagulación están ausentes o su actividad se encuentra reducida. (23)
El mecanismo a través del cual, altas dosis de FVIIra permiten corregir trastornos de coagulación debidos a deficiencias a distintos niveles del sistema, no se conoce del todo. Los mecanismos más importantes parecen ser: la sustitución del FVIIa congénitamente ausente en la iniciación de la hemostasia y la derivación (by-pass) de defectos en el proceso normal de coagulación en ciertos pacientes con coagulopatía. (24)
Se ha demostrado que altas concentraciones de FVII generan trombina, independientemente del FT, a través de una interacción directa con las plaquetas. Las consecuencias de la producción acelerada de trombina, sea dependiente o independiente de FT, incluyen: a) activación de plaquetas, b) generación de IFAT c) activación del factor XIII y d) incremento en la producción de fibrina. (25)
Indicaciones del factor VII recombinante:
Tratamiento de episodios de hemorragia y prevención de hemorragias durante intervenciones quirúrgicas o durante procesos invasivos en pacientes con:
- Hemofilia congénita con inhibidores para los factores de coagulación VIII o IX, o que se espera que tengan una respuesta anamnésica alta con los factores VIII o IX.
- Hemofilia adquirida.
- Deficiencia congénita del factor VII.
- Trombastenia de Glanzmann con anticuerpos contra glucoproteína GP IIb-IIIa y/ó HLA, y con respuestas pasadas o presentes a la transfusión de plaquetas.
El tratamiento con el FVIIra en pacientes con hemofilia tuvo muy buenos resultados, mostrando mayor seguridad y eficacia que con tratamientos tradicionales. (26, 27)
Otras indicaciones:
Sangrado quirúrgico:
- Trauma
- Hemorragia intracerebral
- Hemorragia posparto
- Hepatectomía
- Daño cerebral
- Transplante ortotópico de hígado
- Cirugía cardíaca (de adulto e infantil)
- Cirugía de la columna vertebral
- Cirugía reconstructora de la pelvis
El daño por trauma es la principal causa de muerte entre los 5–44 años y el sangrado es la segunda causa de mortalidad en el daño del SNC. (28-30)
Ejemplos de otras situaciones hemorrágicas críticas son:
- Hemorragia postransplante de médula ósea
- Infecciones víricas hemorrágicas (por ejemplo, dengue)
- Hemorragia aguda por varices esofágicas
- Hemorragia intracerebral
- Lesión por traumatismo cerebral
- Hemorragia posparto
- Quemaduras
- Hemorragia digestiva
- Hemorragia en neonatos pretérminos
- Hemorragia del sistema nervioso central
Altas dosis de factor VIIa pueden corregir trastornos de la coagulación al generar grandes cantidades de trombina en la superficie plaquetaria; estos estudios han llevado a considerar el potencial del FVIIra como un agente hemostático universal. (30, 31)
El uso del FVIIra tiene muy buenos resultados en todas estas afecciones debido al importante papel que juega en el mecanismo de coagulación, pero además tiene como ventaja que evita o disminuye el uso de las transfusiones masivas de glóbulos rojos, lo cual es una variable de riesgo para muchas complicaciones. (31, 32)
Todos estos avances del uso del FVIIra en el tratamiento de un amplio espectro de afecciones con compromiso hemostático se deben en gran parte al estallido de la nueva teoría celular de la coagulación, la cual sustituye un modelo que ha sido empleado tradicionalmente por varias decenas de años a nivel mundial, pero que no dejaba de estar inconcluso en muchos aspectos, dejando grandes interrogantes pendientes que son ahora abarcadas por este nuevo paradigma de la hemostasia. (33)
CONCLUSIONES
En los últimos años se desarrolló un nuevo modelo de la coagulación, el cual propone que esta se activa mediante la interacción de superficies celulares, factor tisular y factor VII en tres fases simultáneas: iniciación, amplificación y propagación, a diferencia del modelo tradicional el cual postula que la coagulación está regulada exclusivamente por una cascada de activación de factores solubles en dos vías independientes.
En este nuevo modelo, también conocido como teoría celular de la coagulación, se propone que las superficies celulares controlan y dirigen el proceso de la hemostasia. La nueva teoría permite un mejor entendimiento de los problemas clínicos observados en los trastornos de la coagulación.
Debido al importante rol que juega el factor VII en el nuevo modelo celular de la coagulación, sobre todo para darle inicio, se está comenzando a utilizar, con muy buenos resultados, para el tratamiento de varias enfermedades que provocan compromiso de la hemostasia.
