INTRODUCCIÓN
La neurofibromatosis tipo I o enfermedad de Von Recklinghausen es una de las enfermedades genéticas que afectan el sistema nervioso, denominada así por su origen común embrionario. Es una enfermedad autosómica dominante, progresiva, de evolución impredecible, que afecta la piel y el sistema nervioso central y periférico; se debe a una mutación del cromosoma 17q11.2, codificante de una proteína supresora tumoral, la neurofibromina.(1,2,3,4)
Probablemente, el primer caso de neurofibromatosis tipo 1 (NF 1) data de hace 2.500 años. Se describen como tumores de consistencia firme, inmóvil, con nudos en su interior, como si estuvieran rellenos de aire, lo que nos haría pensar hoy en día en un neurofibroma plexiforme. Von Recklinghausen fue el primero en utilizar el término «neurofibroma» para describir lesiones donde coexistían elementos neurales y células del tejido conectivo. Esto fue la clave para utilizar el epónimo que lleva su nombre y haber sido considerado el líder de los patólogos del siglo XIX.(5)
La NF1 pertenece al grupo de “enfermedades raras”. La incidencia es de 1/3,000 recién nacidos y la prevalencia se sitúa en torno a 1/50,000 habitantes, presentando una elevada proporción de nuevas mutaciones, aproximadamente el 50 %. La penetrancia es del 100 % con una expresividad variable. Constituye el síndrome neurocutáneo más frecuente.(1, 6)
A pesar de ser una enfermedad sistémica con capacidad para afectar a prácticamente la totalidad de las estructuras orgánicas, es poco conocida, de ahí la importancia del conocimiento de esta enfermedad para poder realizar un diagnóstico precoz así cómo brindar un adecuado asesoramiento genético a las familias afectadas, por lo que se consideró de utilidad realizar la presentación de un paciente de siete años de edad, con el diagnóstico de neurofibromatosis tipo I.
PRESENTACIÓN DEL CASO
Escolar femenina, de siete años de edad, de procedencia rural, hija de padres no consanguíneos, producto de un embarazo que cursó sin alteraciones, parto distócico por cesárea a las 39,2 semanas; peso al nacer de 3800 gr, sin complicaciones posnatales. Con antecedentes personales de asma bronquial con tratamiento regular. Aprendizaje normal. Ingresó en el Hospital Pediátrico Paquito Gonzáles Cueto debido a la presencia de varias manchas en la piel color “café con leche”. Se recogió como antecedente familiar la presencia de neurofibromatosis tipo 1 en la abuela y bisabuela materna, esta última ya fallecida por dicha causa.
Examen físico (datos positivos)
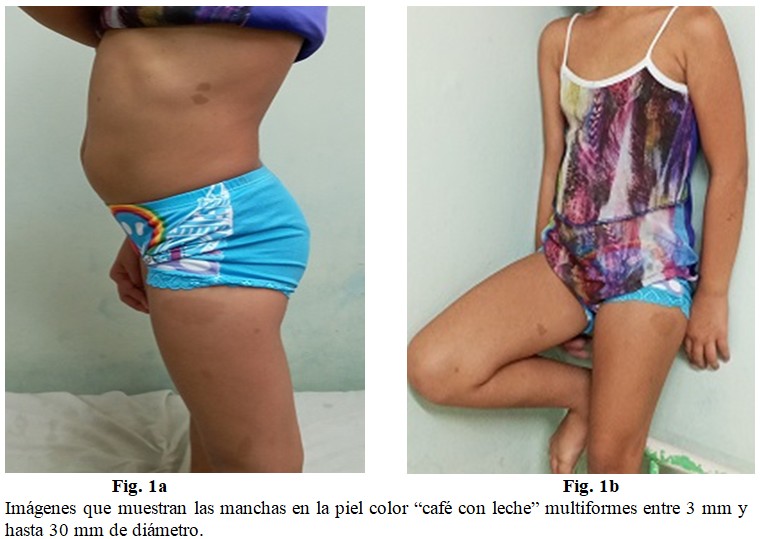
Presencia de varias manchas en la piel color “café con leche” multiformes, de entre 3 mm y hasta 30 mm. (Fig. 1 a y b).
El fondo de ojo arrojó el siguiente resultado: nódulo de Lisch en espesor del iris del ojo izquierdo.
Resultados de los exámenes complementarios
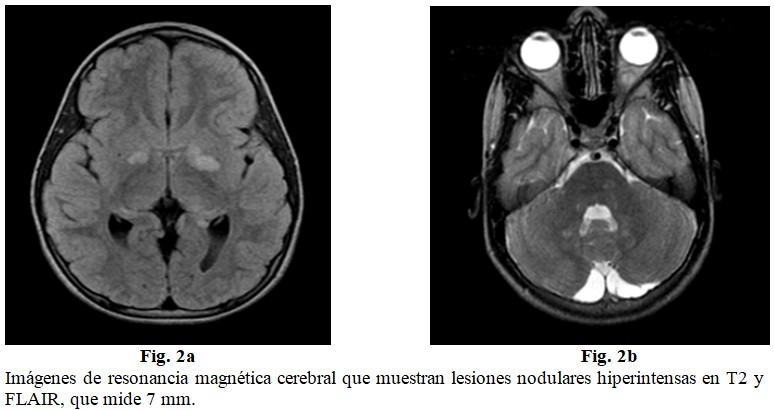
Resonancia magnética cerebral: varias lesiones nodulares que se comportan hiperintensas en T2 y FLAIR, a nivel de los núcleos de la base bilateral y paraventricular izquierda, la mayor de 20 x 11 mm, con excepción de la del nervio óptico izquierdo que se muestra hipointensa en FLAIR que mide 7 mm, no hay efecto de masa ni edema perilesional. (Fig. 2 a y b).
UTS abdominal: negativo.
PEATC y PEV: sin alteraciones.
Test psicométrico: coeficiente intelectual entre 84 y 90 puntos, normal bajo.
Se diagnosticó como neurofribomatosis tipo 1 y se indicó seguimiento por especialistas.
DISCUSIÓN
La NF1 es una enfermedad multisistémica, con síntomas predominantes en sistema nervioso central y periférico, y en piel. Tiene un curso progresivo, con aparición de manifestaciones clínicas heterogéneas, que no siempre están presentes en todos los pacientes. Además, estas manifestaciones presentan una historia natural característica, con aparición de lesiones específicas, típicamente en algunos periodos etarios.(7)
Se caracteriza por una gran variabilidad clínica, no sólo entre diferentes individuos sino también en el propio individuo a lo largo de su desarrollo, lo que dificulta, junto con otros factores, establecer una relación genotipo-fenotipo.(8)
El diagnóstico es clínico; se presenta con neurofibromas en cualquier localización corporal, manchas de color “café con leche”, efélides en axilas e ingles, o neoplasias a nivel del sistema nervioso central (frecuentemente gliomas de nervio óptico), y los pacientes afectados presentan con frecuencia pequeños hamartomas del iris (nódulos de Lisch) o lesiones displásicas óseas.(9)
En el año 1988, el Instituto Nacional de Salud de Estados Unidos estableció como criterios clínicos para el diagnóstico de la neurofibromatosis tipo 1, la presencia de dos o más de los siguientes criterios:(10,11,12)
- Seis o más manchas “café con leche” de más de 5 mm en pacientes prepuberales y de más de 15 mm en pacientes pospuberales.
- Dos o más neurofibromas de cualquier tipo o presencia de un neurofibroma plexiforme.
- Efélides axilares o inguinales.
- Glioma óptico.
- Dos o más nódulos de Lisch (hamartomas del iris)
- Displasia ósea característica (esfenoides o huesos largos con o sin artrosis).
- Familiar de primer grado afectado.
Las máculas “café con leche” constituyen el hallazgo clínico más frecuente y precoz de NF1, y pueden ser congénitas o aparecer a lo largo del primer año de vida (99 % de los casos) y aumentar en número durante la infancia. Estas lesiones no se presentan en el cuero cabelludo, palmas ni plantas, y son de color y tamaño variable, incluso en un mismo individuo; sin embargo, aunque son sugestivas, no son patognomónicas de la entidad. Hasta el 20 % de los niños presentan una lesión hipercrómica aislada, el 4 % tienen dos y menos del 1 % de la población sana tiene más de tres (a estos pacientes se les debe realizar un seguimiento riguroso).(11)
En el caso que se presenta, además de la presencia de las máculas “café con leche”, existe también el antecedente familiar y la presencia de nódulos de Lisch por lo que se planteó el diagnóstico de neurofibromatosis tipo I.
Uno de los hallazgos principales en los niños con NF1 son las hiperintensidades en secuencias T2 en la RM cerebral, hallazgos conocidos como “imágenes brillantes inespecíficas” (IBI o, en inglés, UBO). Se localizan principalmente a nivel de los ganglios basales y cerebelo, aunque pueden estar presentes en tractos ópticos, tronco encéfalo, tálamo, entre otras localizaciones, que se encontraron presentes en este caso.(7, 8)
La mayoría de los pacientes con NF1 tienen un coeficiente intelectual similar al de la población general, aunque hasta un 50-80 % puede tener dificultades de aprendizaje y trastornos de la conducta. Un 30 % de los niños puede presentar algún trastorno del espectro autista, y cerca de un 40 %, un trastorno por déficit de atención e hiperactividad. En este contexto es fundamental realizar un seguimiento estricto del desarrollo de las funciones cognitivas y del desempeño escolar en los niños con diagnóstico de NF1. Se recomienda realizar periódicamente test de coeficiencia intelectual y evaluaciones neuropsicológicas para detectar tempranamente alteraciones cognitivas e iniciar un apoyo escolar diferenciado.(3,6,12, 13,14)
Se ha intentado establecer una relación entre la presencia de hiperintensidades en secuencia T2 de resonancia magnética con la aparición de trastornos de aprendizaje y deterioro cognitivo. La presencia de hiperintensidades a nivel de cerebelo parecería relacionarse con un coeficiente intelectual más bajo y dificultades a nivel de la memoria visoespacial; sin embargo, no existen datos concluyentes.(8)
El test de coeficiencia intelectual que se realizó inicialmente en este paciente resultó ser normal bajo. No se constataron alteraciones de la conducta, trastornos del espectro autista ni trastorno por déficit de atención e hiperactividad.
La incidencia de convulsiones en los pacientes con NF1 es mayor que en la población general debido a la presencia de tumores o infartos del sistema nervioso central. Pueden ocurrir a cualquier edad y, por lo general, se expresan como convulsiones focales. Para que el tratamiento sea exitoso habitualmente se requiere de la combinación de distintos fármacos antiepilépticos o de la resección quirúrgica de la región afectada.(6) En este caso aún no han aparecido eventos convulsivos.
La NF1 puede afectar a prácticamente todos los órganos y sistemas, provocando, además de trastornos del aprendizaje, trastornos oftalmológicos y neurológicos, otros como ortopédicos, cardiovasculares y tumorales. Los tumores malignos son, probablemente, la complicación más temida de la NF1. La implicación de la neurofibromina va a interferir con la proliferación y diferenciación celular y, por lo tanto, aumentará la predisposición al desarrollo de tumores malignos en estos pacientes.(6)
El tratamiento de la neurofibromatosis tipo 1 debe ser individualizado; depende de la localización del tumor, si causa dolor o desfiguración importante, si afecta la función y de su velocidad de crecimiento. Las opciones van desde la observación, la resección quirúrgica parcial o total y el uso de quimioterapia.
La neurofibromatosis tipo I es una enfermedad multisistémica y progresiva, donde los tumores malignos constituyen la complicación más temida; además, poco conocida por el personal médico, con un diagnóstico que se realiza fundamentalmente por las manifestaciones clínicas; por lo que se hace necesario enfatizar en el conocimiento de las mismas para garantizar su diagnóstico desde etapas tempranas de la vida que nos permita un seguimiento oportuno y un diagnóstico precoz de sus complicaciones.
Conflicto de intereses
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Contribuciones de los autores
Conceptualización: Marisleidy Denis Rodríguez, Miguel Alejandro Pulido Gutiérrez, Luis Omar López Hurtado.
Visualización: Thaimí Conde Cueto.
Redacción del manuscrito: Marisleidy Denis Rodríguez, Thaimí Conde Cueto.
Revisión, redacción y edición: Marisleidy Denis Rodríguez, Miguel Alejandro Pulido Gutiérrez.
Financiación
Hospital Pediátrico Universitario Paquito González Cueto. Cienfuegos.
