INTRODUCCIÓN
La poroqueratosis (PQ) es una enfermedad clonal de la queratinización caracterizada por una o más placas atróficas rodeadas por un borde en forma de cresta, clínica e histológicamente distintivo, llamado lamela cornoide. (1) Cinco variantes clínicas de poroqueratosis han sido reconocidas: poroqueratosis clásica de Mibelli (PM), poroqueratosis actínica superficial diseminada (PASD), poroqueratosis palmoplantar diseminada (PPPD), poroqueratosis lineal, y poroqueratosis punctata. (1) También ha sido descrita una variante inusual verrugosa que es localizada en la región glútea y parecida a la psoriasis. (2)
Las lesiones de la PQ clásica fueron descritas por Mibelli, un dermatólogo italiano, en 1893, quien le asignó el nombre de poroqueratosis porque creyó que estas lesiones comenzaban en las glándulas sudoríparas. (3) Una forma diseminada más superficial fue descrita por Respighi alrededor de ese mismo tiempo, y la variante lineal se incluye en este mismo siglo; en 1966 la PASD fue descrita por Chernosky y en 1971 Guss y colaboradores añadieron la PPPD. (4, 5)
La hiperproliferación clonal de los queratinocitos atípicos conduce a la formación de la “lamela cornoide”, que se expande periféricamente y forma el límite elevado entre los queratinocitos anormales y normales. (1) La inmunosupresión local o sistémica puede permitir el desarrollo de clones de queratinocitos atípicos en individuos que están genéticamente predispuestos. (6) Un modo autosómico dominante de herencia ha sido establecido para los casos familiares de todas las formas de PQ. (1) La inmunosupresión puede inducir nuevas lesiones o causar una agudización de las lesiones preexistentes. (1, 2, 5)
La degeneración maligna ha sido reportada en todas las formas de poroqueratosis, con un riesgo de 7,5 % a 11 % determinado en dos revisiones publicadas en la literatura médica. (7, 8) Las hipótesis más aceptadas para el desarrollo de enfermedades malignas cutáneas en el curso de la poroqueratosis son la inestabilidad cromosómica y la reducida vigilancia inmune con sobreexpresión de la proteína p53. (1, 8)
La descripción de un caso familiar de PM ha motivado la realización de este trabajo, que constituye el primer reporte en la provincia y en el país.
PRESENTACIÓN DEL CASO
Paciente de 44 años de edad, de sexo masculino, con antecedentes de hipertensión arterial primaria estadio 2 sin tratamiento, trabajador de la empresa de Comunales, sin contacto con sustancias tóxicas y sin hábitos tóxicos, que acudió al área de atención secundaria para realización de chequeo preempleo. Refirió que desde la infancia poseía lesiones en todo su cuerpo, las cuales fueron aumentando progresivamente en número y tamaño, aunque no le reportaban síntoma alguno. Durante el interrogatorio reveló que varios miembros de su familia (tanto ascendientes como descendientes) poseían manifestaciones dermatológicas con similar comportamiento.
Examen físico
Paciente sin afectación del estado general, afebril.
Aparato cardiorrespiratorio: frecuencia respiratoria: 20 resp/min, murmullo vesicular conservado, ruidos cardiacos rítmicos; tensión arterial: 160/100 mm Hg, frecuencia cardiaca central: 95 lat/min.
En la piel se constataron lesiones en forma de placas anulares, diseminadas en todas las regiones corporales, con respeto de las mucosas; de tamaño y forma irregular, atróficas, sin pelo, anhidróticas, algunas de ellas hiperpigmentadas, con un borde ligeramente hipertrófico que las separa de la piel normal (Fig. 1 A y B).
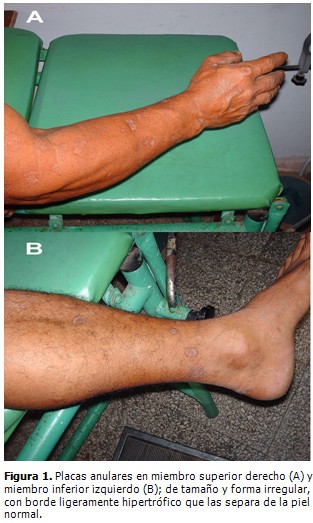
El resto del examen físico fue totalmente normal.
Fondo de ojo: retinopatía grado I.
Los estudios analíticos, electrocardiográficos e imagenológicos, incluyendo ultrasonografía, fueron todos normales.
Seguimiento por consulta de dermatología
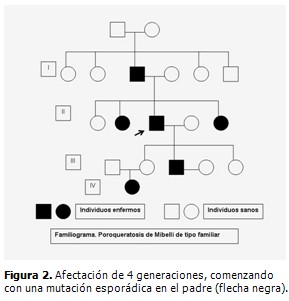
El paciente fue remitido a la consulta de dermatología donde se sospechó la presencia de una genodermatosis. Fueron valorados los demás miembros de la familia, constatándose la presencia de lesiones similares en el padre de 70 años de edad, dos hermanas y un hijo varón de 20 años (Fig. 2). En el hijo las lesiones eran incipientes con predominio de pápulas carmelitas, pequeñas, hiperqueratósicas, diseminadas, algunas de ellas pruriginosas y algunas en forma de placas como las ya descritas. (Fig. 3 A y B)
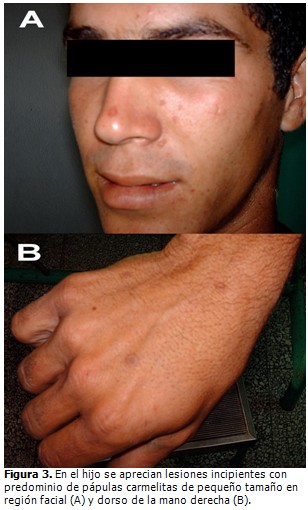
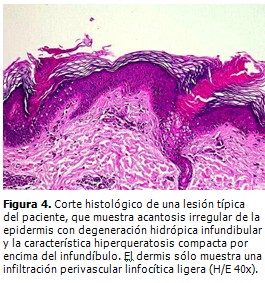
El estudio histopatológico (Fig. 4) confirmó el planteamiento clínico de una PM, con presencia de la “lamela cornoide” como elemento más característico y sin hallazgos de malignidad en los elementos celulares. En la dermis papilar sólo se encontró un infiltrado linfocítico perivascular.
Tratamiento
En un inicio, se empleó como terapéutica el 5-fluorouracilo tópico, con lo que se obtuvo mejoría parcial pero con gran inflamación de las lesiones, por lo que se suspendió para aplicar a los 10 días imiquimod al 5 % en crema, con mejores resultados sobre las lesiones.
DISCUSIÓN
Las PQ son enfermedades raras, sin embargo la PASD es frecuente en algunos sitios como en los Estados Unidos de América; estas entidades son más frecuentes en personas rubias que en los de piel oscura. La PM afecta a los hombres en proporción 2:1 frente a las mujeres, desarrollándose con frecuencia en la niñez. (1-4) En nuestros casos predominaron los varones, todos eran de piel trigueña y debutaron en la infancia.
Las lesiones de PM pueden localizarse en cualquier sitio del tegumento: cara, extremidades, genitales, glúteos, etc.; generalmente permanecen asintomáticas, a veces con prurito discreto durante algunos períodos del año; de manera infrecuente se desarrollan en la adultez y crecen con rapidez, particularmente en sitios afectados por traumas o quemaduras; (1,9) los casos estudiados presentaban las lesiones de forma diseminada y sin ningún antecedente de importancia.
La lesión de la PM se desarrolla como una pápula queratósica, pequeña, de color carmelita brillante, que aparece lentamente y se expande con límites irregulares, hasta formar placas anulares, bordes acordonados y elevados, los cuales pueden observarse hípertróficos o verrugosos y casi siempre tienen un tamaño superior a 1 mm de altura.
Las lesiones pueden ser hiper o hipopigmentadas, con descamación discreta, de centro atrófico, sin pelo, con marcada anhidrosis y con tamaños que varían desde milímetros a varios centímetros. (1, 7, 10) Todas estas características fueron apreciadas en nuestros pacientes.
Entre los factores de riesgo de la PM se señalan la herencia de tipo autosómico dominante, la inmunosupresión, la radioterapia, las quemaduras y la hemodiálisis; (11) de ellos el único identificado fue el factor genético.
El diagnóstico diferencial de la PQ debe realizarse con la psoriasis en placa, el nevus epidérmico lineal verrugosoy el nevus ostial ecrino poroqueratósico. (1, 5, 6)
Por lo general, los estudios analíticos, radiológicos e inmunológicos son normales, (1) como se evidenció en nuestros pacientes; sin embargo, deben practicarse todos los exámenes relacionados con el VIH y las malignidades hematológicas, al igual que las investigaciones relacionadas con la función renal. (12)
El estudio histopatológico es fundamental en el diagnóstico de todas las PQ, y la "lamela cornoide" el hallazgo que confirma el diagnóstico, pero es esencial que el tejido que se obtenga sea de una elevación hiperqueratósica periférica. (1, 7, 8) La lamela cornoide consiste en una fina columna de células paraqueratósicas empacadas dentro de una invaginación epidérmica llena de queratina. Macroscópicamente, la lamela cornoide se presenta en el borde en forma de cresta elevada rodeando a las placas anulares, atróficas, siendo más prominente en la PM. (1, 7, 8)
El estudio histopatológico es de máxima importancia en el diagnóstico y seguimiento de los pacientes, sobre todo en lesiones ulceradas persistentes debido al potencial neoplásico de la PM, ya que se ha demostrado el desarrollo de carcinoma espinocelular con metástasis de evolución fatal en estos pacientes. (13)
El tratamiento de la PQ debe ser individualizado, basado en el tamaño de la lesión, en la localización anatómica, en consideraciones estéticas y en la preferencia del paciente. (1) La protección del sol, el uso de emolientes y la observación de signos de malignidad deben tenerse también presentes. (1, 6, 7, 12, 13)
Se ha demostrado que el 5-fluorouracilo tópico induce remisión de todas las formas de PQ pero debe emplearse solo hasta la aparición de reacciones inflamatorias; se han señalado recurrencias con este tratamiento. (14) También han sido empleados los análogos tópicos de la vitamina D-3 en la PASD durante meses, siendo efectivos en estos casos. Los mejores resultados en el tratamiento de la PM se han obtenido con el imiquimod al 5 % en crema, (15) tal como se demostró en la familia en estudio. Debido al tiempo de evolución, algunos de los miembros de la familia se encuentran bajo estudio y control histológico permanente, con el fin de detectar oportunamente el desarrollo de malignidad.
Se han empleado los retinoides orales en los inmunosuprimidos, los cuales tienen alto riesgo de malignización, (1, 7, 8, 14) y el etretinate, cuya eficacia ha sido ampliamente discutida, al igual que el acitretin, un retinoide de segunda generación y que según algunos autores (1, 16) son útiles en la PQ lineal y en la PM. El tratamiento quirúrgico es esencial en la PQ que presenta transformación maligna. (1, 11, 13)
La PM es una entidad importante, ya que en su diagnóstico diferencial se integra el enorme grupo de las manifestaciones dermatológicas de los procesos internos, incluídos los fenómenos paraneoplásicos. Su manejo es esencial, pues requiere atención multidisciplinaria dado el hecho de que los individuos de varias generaciones pueden ser afectados.
